
Abstract
As 3D printing becomes more common and the technique is used to build culture platforms, it is imperative to develop surface treatments for specific responses. The advantages of aminating and oxidizing polystyrene (PS) for human mesenchymal stem cell (hMSC) proliferation and osteogenic differentiation are investigated. We find that ammonia (NH3) plasma incorporates amines while oxygen plasma adds carbonyl and carboxylate groups. Across 2D, 3D, and 3D dynamic culture, we find that the NH3- treated surfaces encouraged cell proliferation. Our results show that the NH3-treated scaffold was the only treatment allowing dynamic proliferation of hMSCs with little evidence of osteogenic differentiation. With osteogenic media, particularly in 3D culture, we find the NH3 treatment encouraged greater and earlier expression of RUNX2 and ALP. The NH3-treated PS scaffolds support hMSC proliferation without spontaneous osteogenic differentiation in static and dynamic culture. This work provides an opportunity for further investigations into shear profiling and coculture within the developed culture system toward developing a bone marrow niche model.
Impact statement
Surface treatment can be leveraged to enhance human mesenchymal stem cell response when transitioning polystyrene from a 2D to a 3D culture substrate. Understanding how the underlying surface chemistry influences the adhered cells could help build complex culture environments, with multiple cell types and work toward more biomimetic models of the bone marrow niche. Toward this goal, it is imperative to establish how the cells respond under static and dynamic culture and ensure the scaffolds support osteogenesis under typical induction conditions.
Introduction
As 3
A variety of PS culture substrates are on the market, suiting the needs of biologist, engineers, and researchers across disciplines. However, the base material lacks the surface chemical functionality to best facilitate protein adhesion and cell spreading.3,4 To address this shortcoming, tissue culture polystyrene (TCPS) undergoes an oxygen (O2) plasma treatment to generate a surface conducive to cell adhesion and proliferation,5,6 although several plasma compositions have been investigated. 7 The competing chemical, thermodynamic, and electrostatic forces at the surface makes quantifying every interaction difficult, but understanding the link between chemistry, surface energy, and cell response provides a good summary for building culture systems.
Understanding the impact of surface properties and proteins on cell-substrate interactions within the culture environment is essential in tissue engineering and regenerative medicine applications. The complex interplay between substrate properties and adhered proteins is crucial to achieving populations with expected differentiation capacity in pluripotent stem cells. 8 PS culture surfaces grafted with poly(allylamine) have been shown to support human mesenchymal stem cell (hMSC) adhesion and chondrogenesis, a phenomenon not seen with poly(acrylic acid) grafted surfaces. 9 Carboxylic acid-modified surfaces have been shown to enhance chondrogenic differentiation,10–12 and amines have been shown to enhance calcification and osteogenic marker expression.13,14 Positively charged poly(acrylic acid)-modified surfaces have been found to increase hMSC proliferation and support osteogenic differentiation. 15 Further biomimicry can be gained by transitioning to 3D substrates, with hydrogels continuing to demonstrate cell differentiation trends. 16 Linking chemical influences to 3D culture systems serves the research community by mimicking the natural environment of adherent cell culture and provides a tool to investigate hMSC processes under flow.
Continuing to develop complex, 3D culture systems serves the research community well, better mimicking the natural cell culture environment. Specifically, transitioning PS from a 2D to a 3D platform capable of supporting dynamic culture would provide a unique tool to investigate hMSC processes under flow 17 while growing on a standard culture substrate. The assumed biomimicry afforded by dynamic 3D culture by rapid modeling, tuning, and testing various physiological stressors, including fluid flow, 18 compression, 19 O2 content, 20 cellular distribution, 21 cellular patterning, 22 and overall geometry, 23 justifies continued inquiry. Developing defined, easily producible, and predictable culture platforms for dynamic culture provides an opportunity to improve culture practices broadly.
Along this rational, we sought to develop a plasma treated, 3D printed PS scaffold and bioreactor system for hMSC culture. Particularly, we sought to understand how the surface treatment influences both proliferation and osteogenic differentiation. To accomplish this goal, we first characterized the effect of our surface treatment on PS and determined the saturation point of our plasma treatment system. Next, we sought to understand proliferation and osteogenic differentiation with and without induction media in 2D and 3D culture conditions. Finally, we investigated the performance of our scaffold within a perfusion bioreactor. Overall, this study focused on the impact of surface treatment on hMSC expansion and differentiation, working to advance PS to a 3D culture substrate.
Materials and Methods
3D scaffold fabrication
A 3D Bioplotter (EnvisionTEC GmbH, Gladbeck, Germany) was used to fabricate scaffolds, as described. 24 In brief, a sugar-glass support material (EnvisionTEC GmbH) was printed and used as a support substrate with PS (>50,000 MW atactic flakes; Polysciences, Inc., Warrington, PA) extruded to fabricate 15 mm diameter scaffolds of five layers with a center-to-center fiber distance of 1 mm extruded at 155°C through a 0.4 mm ID needle tip, at 900 kPa, at 3–5 mm/s. Each layer in the scaffold was set at an 85.5° offset to the previous layer, creating a structure which did not repeat over the scaffold height. At the completion of prints, support material was solubilized with ddH2O cleaned in 100% ethanol (EtOH) for 30 min and air dried.
Surface activation through plasma treatment
Test samples were activated using a Femto Diener Electronic vacuum plasma system (Ebhausen, Germany) (typically below 150 Pa). The entire chamber was filled with test gases O2 (Roberts Oxygen, Gaithersburg, MD) and ammonia (NH3; Matheson, Bernards Township, NJ) added at 10 SCCM and system power set to 80 W. Settings were found to produce consistent plasma activation and chamber filling. The chamber was evacuated and flooded two times before activation. Six-well nontreated (NT) dishes were used for 2D experiments and 3D printed scaffolds for 3D experiments. Samples were sterilized under UV light for at least 20 min before use in cell studies. Experiments were generally performed at least the day following treatment.
Surface characterization through water contact angle
Water contact angle was measured using Drop Shape Analysis (First Ten Angstroms, Inc., Portsmouth, VA) on ∼3 μL drops of deionized water dispensed from a vertically mounted syringe. The pendant droplet, viewed through a high-resolution camera (Zoom 6000 Lens System; Navitar, Rochester, NY), was lowered to touch the surface and detach from the syringe. Five measurements were taken of each surface.
Chemical surface property assessment through X-ray photoelectron spectroscopy
Quantitative spectroscopic measurements using a Kratos Axis Ultra delay-line detector (Kratos Analytical Ltd, Manchester, UK) used in hybrid mode with a monochromatic Al Kα1, 2 X-ray source (hν = 1486.6 eV) determined sample elemental distribution and bonding stoichiometry with survey scans (0–1100 eV, resolution of 1 eV) and high-resolution scans (pass energy of 40 eV, resolution of 0.1 eV). A Gaussian-Lorentzian distribution with a full width at half maximum constraint of ∼1.7 eV was used for peak fitting with three spectra per sample. Charging was accounted for with a low energy electron flood-gun with a filament current of 1.8 A and charge bias of 2.5 V. Differential charging on the surface was not suspected based on a lack of significant peak broadening. Binding chemistry was identified by deconvolving peaks into components based on the overall peak shape.
Structural characteristics of 3D printed constructs through microcomputed tomography
Samples were scanned by μCT (SkyScan 1272; Bruker, Kontich, Belgium) to generate 3D models of the printed constructs following established protocols.25–27 In brief, three samples were imaged using 180° rotation at 40 kV and 220 mA. A resolution of 8 μm/pixel was used with an Al 0.25 mm filter. Raw images were reconstructed, sliced, and analyzed using NRecon and CTAn software packages (Bruker, Billerica, MA). Data were then imported into LabView (National Instruments, Austin, TX) with 10 different longitudinal slices used to measure the maximum feret diameter, Waddel disk diameter, and spacing of fibers (n ≥ 13/scaffold).
Cell culture
hMSCs (RoosterBio, Frederick, MD) were expanded in RoosterNourish media (RoosterBio). Cells were maintained in a humidified 37°C incubator with 5% CO2. To seed studies, culture flasks were washed with pH 7.4 phosphate-buffered saline (PBS) and lifted with 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) (ThermoFisher, Watham, MA) and neutralized with media. Passage 2–5 hMSCs were used from maintained banks and treated as passage 1 upon receipt. Cells were passed into Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 0.1 mmol/L minimum essential media nonessential amino acids, and 100 U/mL penicillin—100 μg/mL streptomycin (ThermoFisher) at the start of cell studies. This media was used as the growth media for all studies.
Osteogenic media contained 1 mmol/L sodium pyruvate (ThermoFisher), 100 nmol/L dexamethasone, 50 μg/mL ascorbic acid 2-phosphate, and 10 mmol/L β-glycerophosphate (Sigma-Aldrich, St. Louis, MO), as described. 28 Cells were seeded at 5,000 cells/cm2 for 4 h and then sufficient growth media was added to cover samples. Cell counting accomplished using the Trypan Blue (Sigma-Aldrich) exclusion method with a hemocytometer. Media was changed at each time-point for 10-day studies, with induction media added at day 1, as appropriate, and twice per week for 3-week studies.
DNA quantification
Cells were dissociated to understand usable cell quantities from three samples with Trypsin and 0.25% EDTA. Samples were rinsed with PBS and added to lifted cell suspension. Cells were pelleted and resuspended in PBS with DNA isolated following the procedures for DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Isolated DNA was mixed with the diluted dye in technical triplicate and compared to a standard DNA ladder, following protocols for Quant-iT PicoGreen (ThermoFisher). Fluorescence measurements were read on a M5 SpectraMax plate reader (Molecular Devices, San Jose, CA) with an excitation wavelength of 490 nm and emission read at 538 nm. DNA proliferation was evaluated from the D1 timepoint.
Reverse transcriptase polymerase chain reaction
Sample RNA was isolated using TRIzol (ThermoFisher) phase separation techniques. In brief, three samples were washed with a single volume of TRIzol solution and gently mixed to generate a pooled sample. TRIzol solution was transferred to microcentrifuge tubes, chloroform added, and vigorously mixed. The mixture was centrifuged at 12,000g for 15 min at 4°C with the aqueous phase removed. Seventy percent EtOH was added to account for 35% of the total solution volume and transferred to an RNeasy mini column (Qiagen). Complete RNA isolation followed kit instructions.
cDNA conversion was completed using a High Capacity cDNA Archive Kit (ThermoFisher) following manufacturer's procedures. ΔΔCT gene expression was completed in technical triplicate, comparing TaqMan probes for genes of interest (RUNX2, ALP, and OPN) to GAPDH (endogenous control) with Universal Master Mix (ThermoFisher). A 7900HT RT-PCR system (Applied Biosystems, Foster City, CA) cycled reaction vessels as follows: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. NT day 1 growth media samples were used for normalization.
Alizarin Red S staining, imaging, and solubilized stain quantification
At least two samples were washed with Hank's buffered salt solution (Sigma-Aldrich), fixed with 4% buffered formaldehyde 1% sucrose solution for 15 min, and rinsed three times with PBS. Calcification was stained with 40 mM pH 4.1 Alizarin Red S (ARS; Sigma-Aldrich) as previously described.29,30 In brief, samples were washed four times with PBS and imaged (Nikon, Tokyo, Japan). To solubulize the stain, samples were incubated for 30 min at room temperature in 10% (v/v) acetic acid. Samples were mechanically detached and supernatant transferred to microcentrifuge tubes (3D scaffolds transferred as well), vortexed, and overlaid with mineral oil. Samples were heated at 85°C for 10 min, cooled on ice, mineral oil removed, and centrifuged at 15,000g for 15 min. The supernatant was transferred and neutralized with 10% (v/v) ammonium hydroxide with absorbance at 405 nm determined in technical triplicate on a plate reader (M5 SpectraMax; Molecular Devices). Absorbance was normalized to NT growth media samples.
Bioreactor fabrication and assembly
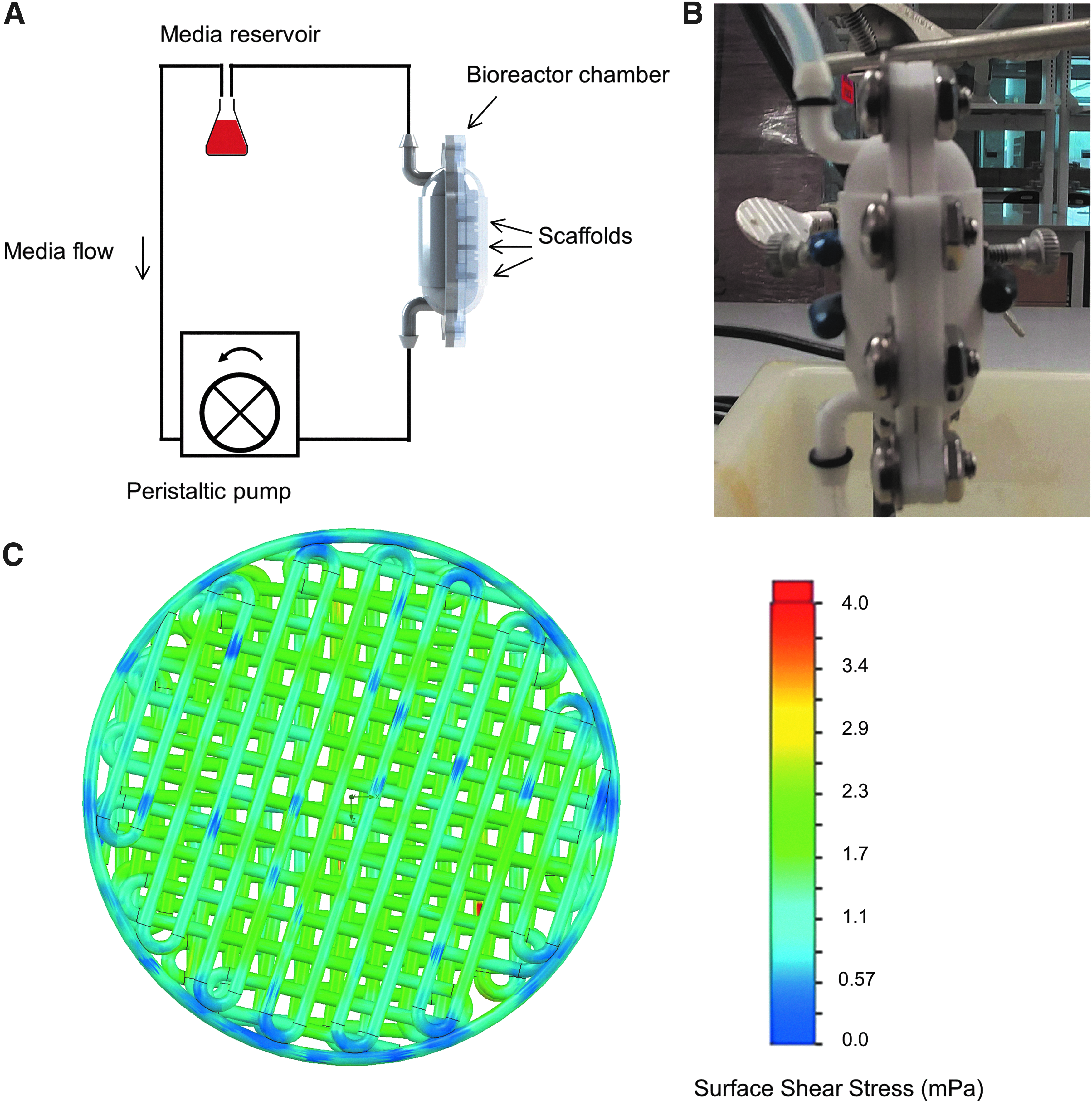
Bioreactor chambers were designed in SolidWorks (Dassault Systemes, Velizy-Villacoublay, France) with STL files exported, aligned, and support structures generated (Magics, Materialise, Leuven, Belgium), and fabricated with Eshell 300 on a Perfactory 4 DSP printer (EnvisionTEC) following manufacturer's procedures. Excess resin was removed from objects by soaking in 99% isopropyl alcohol (IPA) for ∼5 min, rinsing with IPA, and blowing the objects dry with air. This process was repeated until complete resin removal. Support structures were removed and chambers fully cured with up to 2000 flashes of broad-spectrum UV light (Otoflash; EnvisionTEC). Chambers were cleaned in 100% EtOH for 30 min and sterilized under UV light in fresh 100% EtOH for 20 min, and rehydrated as described. 31 Chambers were stored in fresh sterile PBS at 4°C until use and cleaned between uses.
Fourteen-gauge pump tubing was connected to cut 1/8″ ID platinum-cured silicone tubing by 1/16″ × 1/8″ Kynar connectors and fed into 2-hole #5 stoppers (Cole-Parmer, Vernon Hills, IL). Open connections to the media reservoir and bioreactor chamber were covered in aluminum foil. One hundred twenty-five milliliter Erlenmeyer flasks were used as media reservoirs. 50 A durometer silicone sheet of 0.5 mm thickness (Small Parts) was cut using an Epilog laser fusion M2 laser engraver to make gaskets. Stainless steel hardware was used to seal chambers. These components were autoclaved.
Following overnight static cell seeding, three scaffolds containing seeded cells were placed in each chamber, sealed, closed, and attached to Erlenmeyer flasks filled with 50 mL of growth media. A Masterflex L/S digital drive multichannel pump (Cole-Parmer) set at 3.4 mL/min was used to drive flow within a cell incubator. Flow rate was determined by simulating the bioreactor assembly in SolidWorks Flow Simulation to determine a shear stress approximately an order of magnitude lower than typically used to induce osteogenic differentiation (1.5 mPa surface shear stress).32,33 At media changes, media was removed from the flasks and fresh 50 mL was added. At time points, chambers were disassembled and scaffolds processed.
Confocal imaging
Ten day samples were washed with Hank's buffered salt solution (Sigma-Aldrich), fixed with 4% buffered formaldehyde 1% sucrose solution for 15 min, and washed three times with PBS. Permeabilization was performed for 5 min with a 300 mmol/L sucrose, 50 mmol/L sodium chloride, 6 mmol/L magnesium chloride, 20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 0.5% Triton-X-100 solution. Actin was stained with 2.5% AlexaFluor 594 phalloidin (ThermoFisher) in PBS and counterstained with 1 μg/mL diamidino-2-phenylindole. A SP5 X Leica Microsystems confocal microscope (Buffalo Grove, IL) captured Z-stacks. ImageJ projected images along the imaging axis and generated 3D renderings.
Statistical analysis
Statistical significance was determined through a one-way two-sided analysis of variance (ANOVA) at the 95% confidence interval (p < 0.05) using the Games-Howell comparison method in Minitab 18 (Minitab, Inc., State College, PA). Data are presented as mean ± standard deviation with replicates stated in methods and figure captions.
Results
NH3 plasma treatment successfully aminated surfaces
We sought to define surface properties which translate from 2D to 3D in creating culture environment amenable for hMSC proliferation with tunable osteogenic response. We found that the plasma treatment saturated (i.e., no further chemical modification) after 5 min for O2 (21.1 ± 1.8% O2) and NH3 (12.1 ± 0.4% nitrogen) (Fig. 1A–C). This plasma treatment exposure was used for all remaining experiments. The increases in surface O2 with the O2 treatment and surface nitrogen on the NH3-treated surface were statistically significant (p < 0.05, Fig. C).
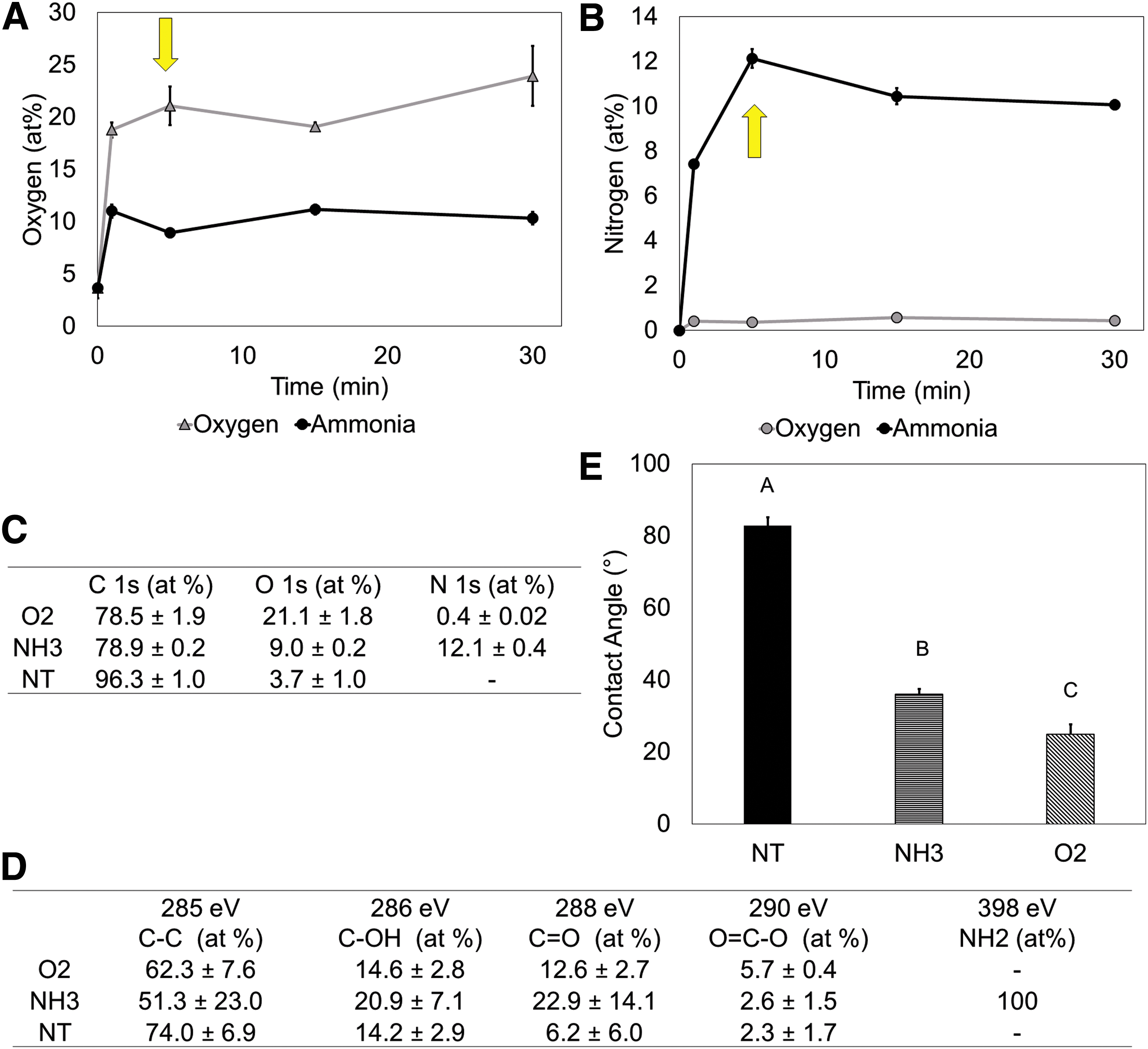
Characterization of surface treatment.
High-Resolution X-ray photoelectron spectroscopy spectra revealed that nitrogen species on the NH3-treated surfaces were predominantly amines (Fig. 1D). Limited oxidation was present on the native NT surfaces (3.7 ± 1.0% O2). Surface treatments significantly decreased the water contact angle for both NH3 (36.0 ± 1.4°) and O2 (24.9 ± 2.9°) surface treatments, consistent with increased surface energy through plasma surface activation (p < 0.05, Fig. 1C).
Surface amines facilitated proliferation and osteogenic differentiation in 2D cultures
Over the course of 10 days, it was observed that generally the NH3-treated surface promoted greater hMSC proliferation compared to the O2-treated or NT-treated surfaces (Fig. 2A). Throughout the study, the greatest amount of DNA was present on the NH3-treated surfaces. It is interesting to note that, while the quantified DNA is similar at the D1 time point, statistically significant differences were observed after 10 days. The DNA content present on the NH3-treated surface (0.63 ± 0.03 μg) was greater than the NT (0.50 ± 0.01 μg) or O2 surfaces (0.53 ± 0.07 μg) (p < 0.05).
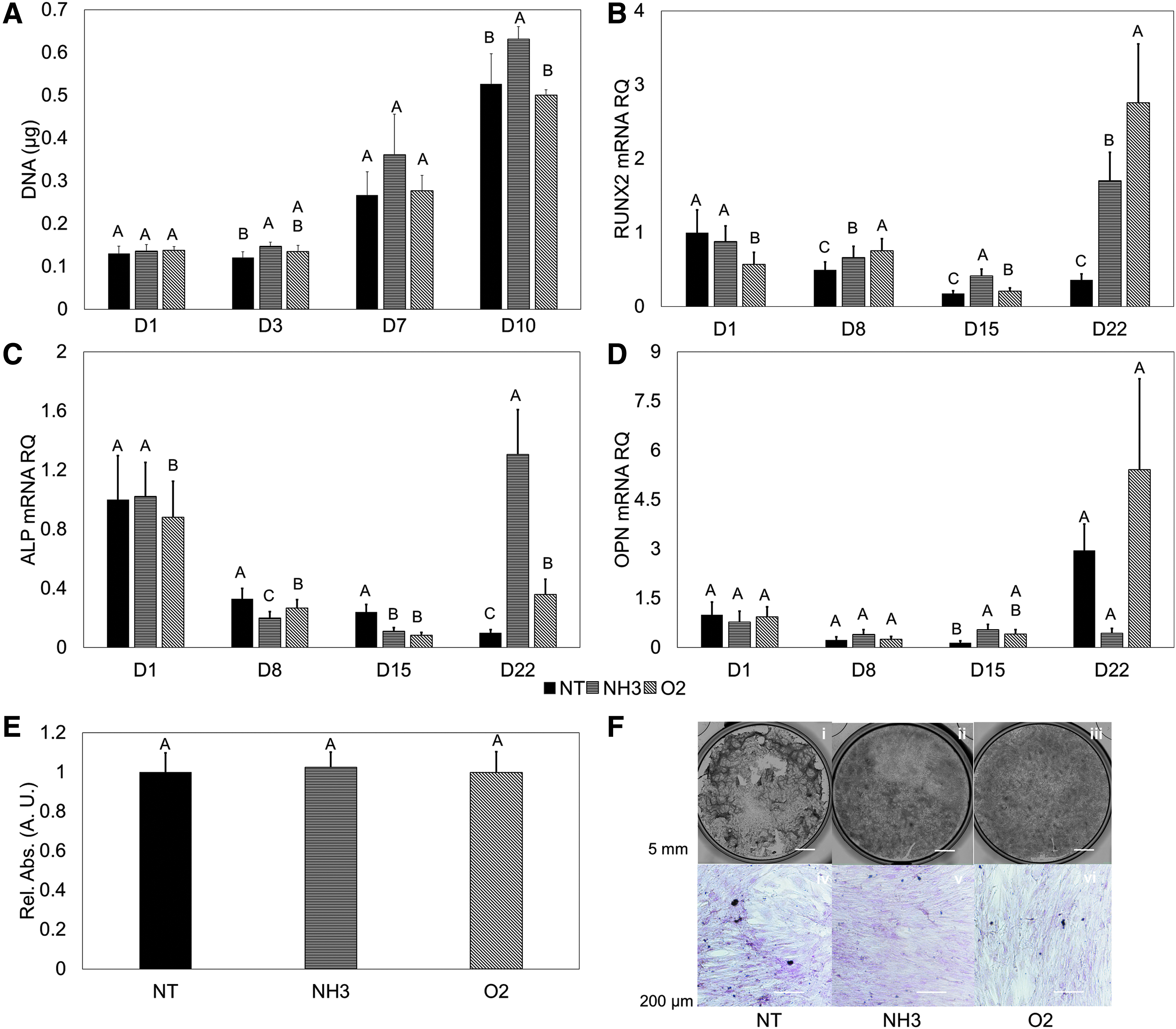
hMSC response in 2D without osteogenic media.
Over a 22-day study, early osteogenic markers (RUNX2 and ALP) were generally downregulated. At the 22-day time point, RUNX2 began to have upregulation. The highest expression was observed on the O2-treated surface, followed by the NH3, and NT surfaces (p < 0.05, Fig. 2B). The only upregulation of ALP expression was observed on the NH3-treated surface at the 22-day time point (p < 0.05, Fig. 2C). OPN saw increasing expression only on the NT- and O2-treated surfaces over the course of the study. Where the greatest differences would be expected, at the final time point, no statistical differences were found (p > 0.05, Fig. 2D). Similarly, ARS staining was performed after 22 days. Semiquantitative assessment determined no statistical differences between cells grown on these surfaces (p > 0.05, Fig. 2E), with little characteristic bright red staining present (Fig. 2F).
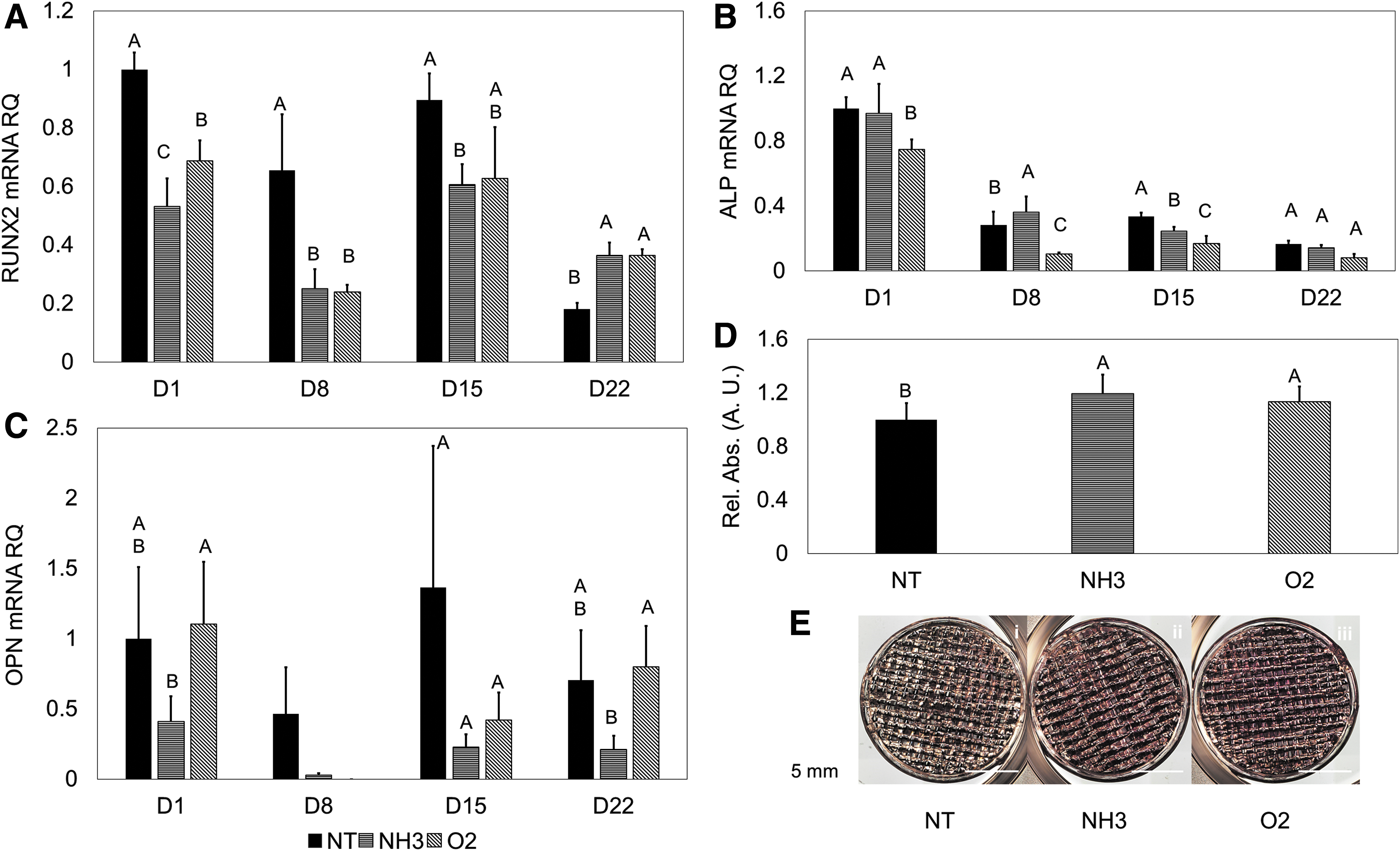
When osteogenic media was used, characteristic differentiation traits were observed. RUNX2 expression increased up until the 8- and 15-day time points for hMSCs on the NH3 surfaces, with expression decreasing at the 22-day time point (Fig. 3A). hMSCs on the O2 surfaces showed continually increasing expression of RUNX2 (Fig. 3A). Peaking expression of ALP between 8 and 15 days was observed for hMSCs on all test surfaces (Fig. 3B). Greater expression of ALP was seen for hMSCs on the NT and O2 surfaces compared with the NH3 surface. Downregulation was observed at the 22-day time point for the NH3 surfaces (p < 0.05). OPN expression generally increased on the NT and NH3 over the time course, with the greatest expression observed on the NH3 surface at 22 days (p < 0.05, Fig. 3C), although values were lower compared with the growth media condition. Characteristic red staining was observed with ARS on all test surfaces, with significantly more solubilized stain present with the O2 treatment (p < 0.05, Fig. 3D, E).
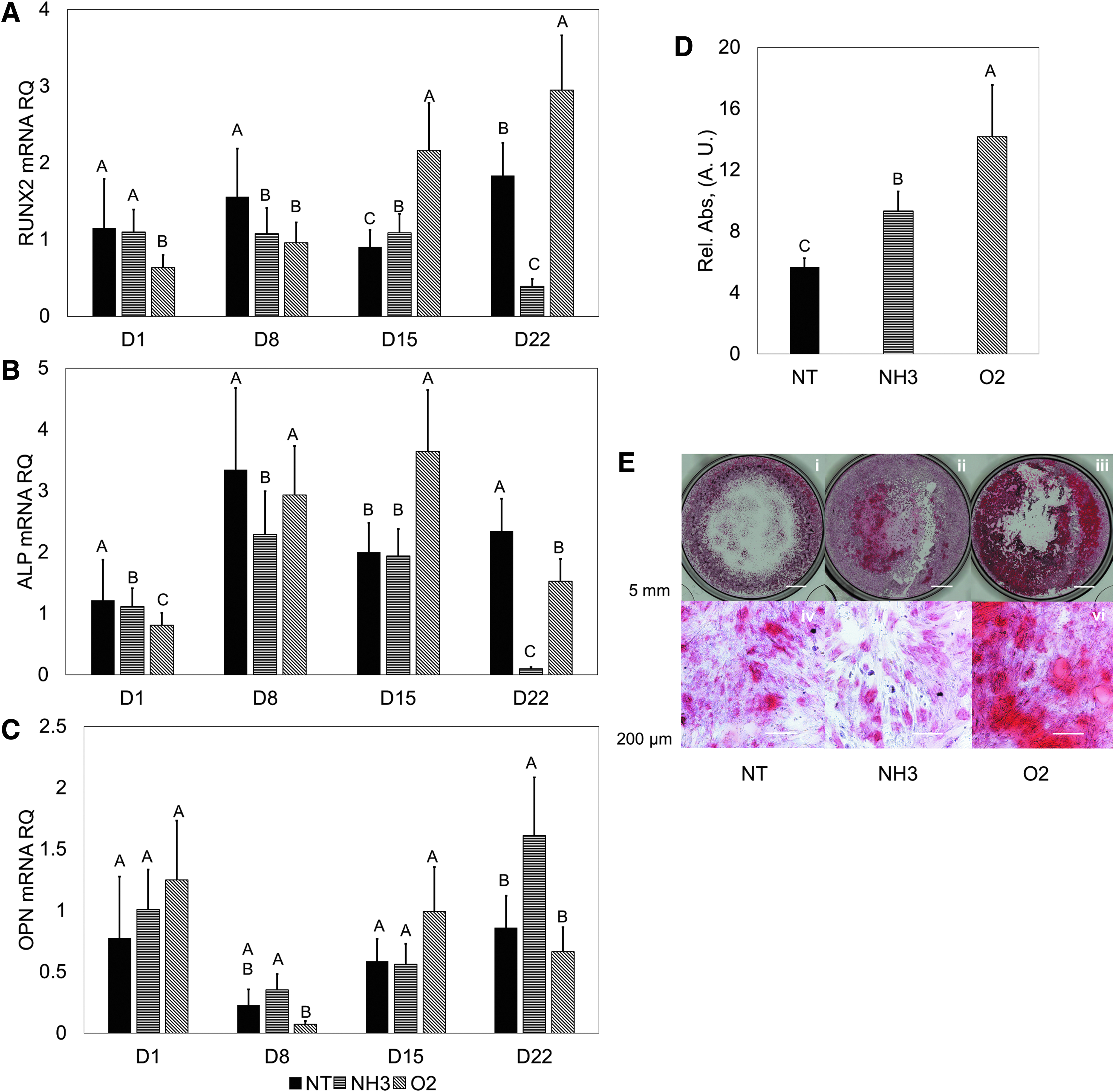
hMSC response in 2D with osteogenic media.
Effect of surface amination persists when translated from 2D to 3D culture
Having established our treatment method facilitated hMSC expansion and osteogenic differentiation in 2D, we then investigated using short (five layer, ∼10.5 cm2 surface area) scaffolds. Fabricated scaffolds were found to have open internal geometry and the fabricated structures were very near the intended design (Supplementary Fig. S1).
Under static conditions, we saw that the treated scaffolds (O2 and NH3) had consistent proliferation over the time-course (Fig. 4A). The NT scaffolds appeared to have inconsistent proliferation: little change was observed between days 4 and 7 and increases of 8.7 × and 3.9 × from days 1 and 3 and days 7 and 10, respectively (Fig. 4A). Characteristic hMSC spreading on and between the fibers on the treated scaffolds was observed, similar to behavior imaged on flat TCPS (Fig. 4B, Supplementary Fig. S2). Spreading was seen within the complex 3D surface on the NH3-treated scaffold through 3D rendering (Fig. 4B, Supplementary Fig. S2). On the NT scaffolds, more crowded and overlaid cells were observed (Fig. 4B, Supplementary Fig. S2).
hMSC growth in 3D without osteogenic media.
Similar to the 2D surfaces without osteogenic media, downregulation of RUNX2 was observed across the time course on all surfaces. hMSCs had greater expression of RUNX2 on the NT scaffold than on the NH3-treated scaffold during the 8 and 15 day time points (p < 0.05, Fig. 5A). ALP was consistently downregulated during the entire time-course Fig. 5B). OPN expression did not see consistent increasing expression (Fig. 5C). Quantified solubilized ARS stain showed statistically greater calcification on the NH3- and O2-treated scaffolds compared with the NT scaffold, and characteristic red staining was confirmed visually (p < 0.05, Fig. 5D, E).
hMSC expression in 3D without osteogenic media.
When osteogenic media was used to direct differentiation, the seeded hMSCs began to undergo osteogenic differentiation. Upregulation of RUNX2 was observed during the time course, with characteristic peaking between days 8 and 15 (Fig. 6A) of the hMSCs on the surfaces. Particularly, the greatest expression was seen on the NH3-treated scaffolds on days 8 and 15 (p < 0.05, Fig. 6A). Similar expression trends during the time-course were seen with ALP expression, with peaking expression occurring between days 8 and 15. Of note, the NH3-treated scaffolds had earlier peaking expression than NT or O2 scaffolds (Fig. 6B). The late osteogenic marker OPN showed the greatest expression at the final time-point in the NT-treated scaffolds, where one would expect continued increasing expression over the time course (p < 0.05, Fig. 6C). Solubilized ARS stain showed no significant differences between the treatments at 22 days (Fig. 6D), with characteristic red staining observed on the scaffolds (Fig. 6E).

hMSC response in 3D with osteogenic media.
Surface amination supported expansion without differentiation in 3D culture under flow
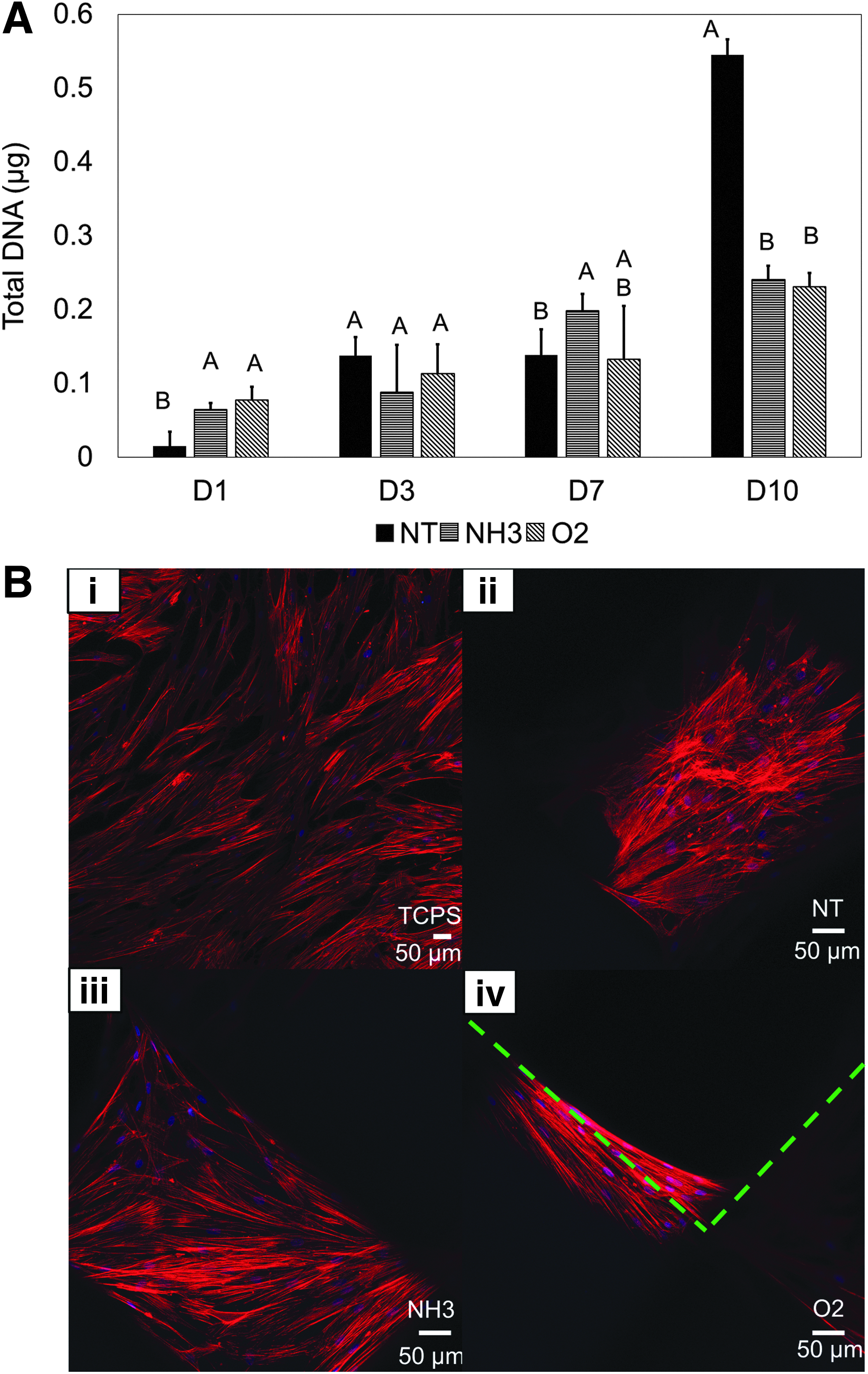
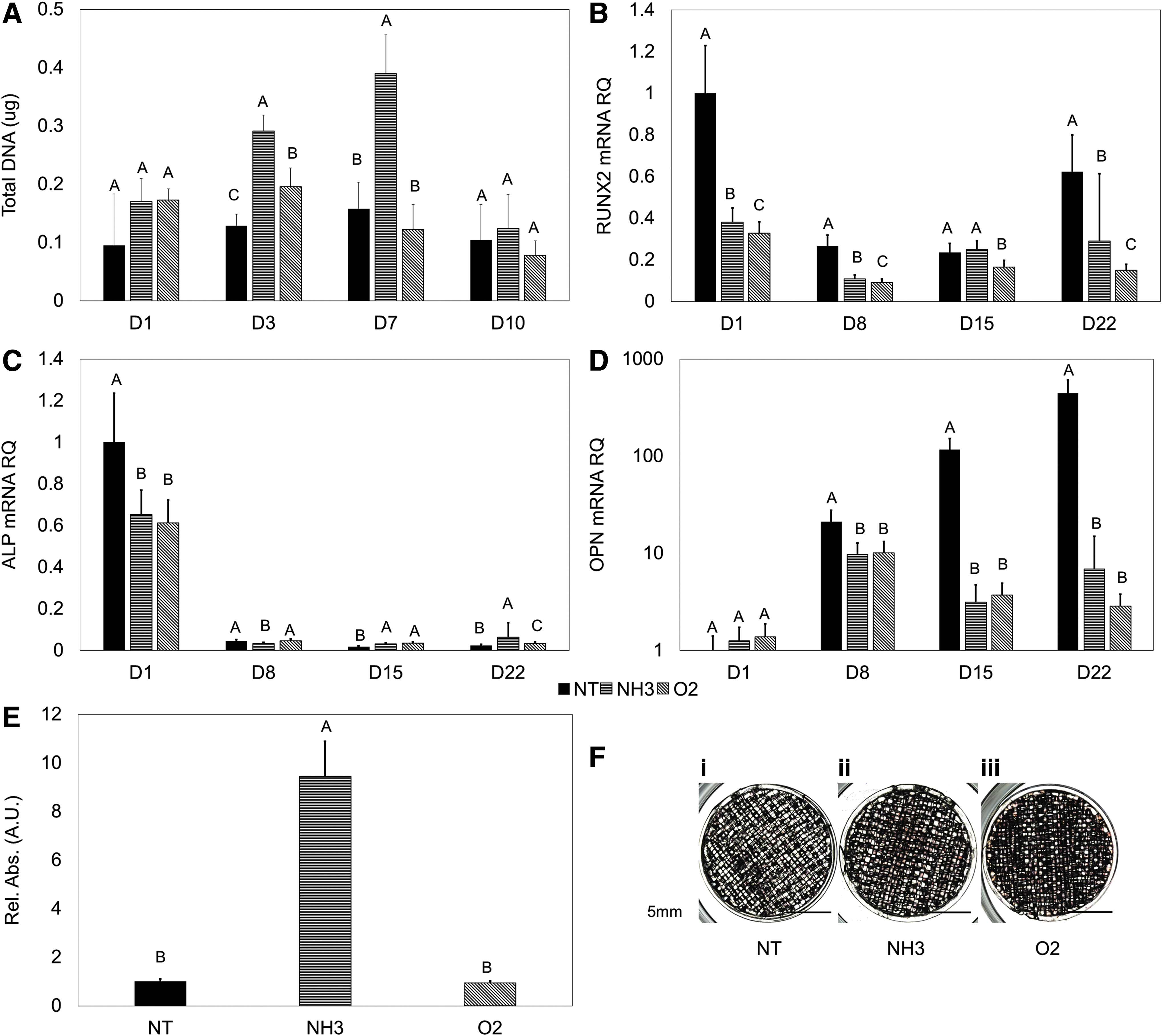
Moving our scaffolds into the bioreactor (Fig. 7A–C) resulted in consistent proliferation for at least 7 days. Particularly, the NH3-treated scaffolds contained the most DNA at days 3 and 7 time (p < 0.05, Fig. 8A). Comparatively, the NT- and O2-treated scaffolds did not appear to support continued dynamic proliferation. Spontaneous differentiation was not apparent on the treated surfaces. hMSC RUNX2 expression generally decreased over time, similar to the static 2D and 3D conditions (Fig. 8B). ALP expression was downregulated across all time-points and treatments (Fig. 8C).
Bioreactor design and shear stress determination.
hMSC response in 3D dynamic culture.
Without added chemical signals to initiate differentiation, the NT-treated surfaces experienced significantly greater, continually increasing OPN upregulation while under flow (p < 0.05, Fig. 8D). OPN expression was seen on the NH3 and O2 scaffolds as well, with no statistical difference between these two groups (p < 0.05, Fig. 8D). NH3 scaffolds were seen to have relatively higher ARS stain after 22 days (p < 0.05, Fig. 8E), but little total calcification observed on all groups (Fig. 8F).
Discussion
As culture systems utilize 3D printing to recapitulate human body stems, focus on the base substrate is warranted. For example, surface properties can influence how hMSCs respond in culture in the development of targeted substrates for tissue engineering applications. Particularly, we sought to establish how hMSCs respond to osteogenic stresses to define an in vitro bone marrow niche model. In the pursuit of a self-contained bioreactor model, we built up from the culture substrate and investigated how hMSCs respond to the culture conditions. As part of this, it remains pertinent to understand both how hMSCs expand and respond to osteogenic stresses.
We began by characterizing treatment surfaces and found vacuum plasma treatments significantly reduced surface carbon (p < 0.05). This is consistent with an extensively modified surface and a greater proportion of functional groups detected, which work with proteins to facilitate cell adhesion and spreading.34,35 The slight surface O2 on the NT surface appeared as a few different species, but with greater oxidation after plasma treatment (p < 0.05), a greater fraction of these chemical species is likely present. The incorporation of O2 and nitrogen outside of the intended treatment (e.g., O2 in the NH3 treatment) likely comes from reacting with the ambient air. The NH3 treatment produced a surface with nitrogen primarily bound as amines, and with water contact angles closest to the range (40°–60°) previously shown to best facilitate cell adhesion. 36 The O2-treated surfaces had lower water contact angles (p < 0.05). Cumulatively, these results demonstrate three distinct surfaces to investigate the influence of surface properties on hMSC response under different culture conditions.
To verify functionality of the constructs, we first investigated hMSC proliferation. The selected scaffold geometry sought to gain inspiration from trabecular bone geometry while creating an object which could be reliably printed. Trabecular bone has a highly porous (9–26% bone volume/tissue volume) structure with trabecular thickness between 120 and 219 μm and trabecular spacing between 667 and 712 μm. 37 The intended design with 400 μm fibers and 600 μm spacing without a repeating layer structure over the scaffold height allows for fluid to penetrate throughout the scaffold without narrowing flow to individual channels as would be seen in a highly structured pattern.
Consistent across the static culture conditions (2D, 3D), aminated surfaces supported hMSC proliferation without spontaneous osteogenic differentiation. Similar results have been previously observed. 38 Doubling times of bone marrow derived hMSCs have been observed to be as high as 2.5 days and higher for hMSCs from other sources. 39 In this study, the doubling time was 3–4 days, within the range of reported doubling times for MSCs (e.g., up to 5 days). 39 We attribute the improved hMSC response on the NH3 treated surfaces and scaffolds to a likely combination of suspected positive surface charge, moderate surface energy, and decreased surface carbon content leading to appropriate protein conformation at the surface.40–45
In addition, we observed that seeding media was rapidly drawn into plasma activated scaffolds, where the NT scaffolds allowed the seeding media sit on top and slowly be drawn into the scaffold bulk during seeding. This could be potentially contributed to poor cell distribution across the NT scaffolds and result in crowding. In addition, the change in geometry could impact how the hMSCs respond and grow on these surfaces, especially having first been accustomed to 2D culture. The data collected suggest that the bulk of the treated objects were activated and allowed hMSCs to interact, adhere, and grow throughout.
We intended to understand how our scaffolds and treatments influence osteogenic differentiation. Geometry of scaffolds can influence hMSC fate,46,47 with the research goal to reduce spontaneous osteogenic differentiation. Two early and two late osteogenic markers were surveyed to ensure that our treatments produced culture platforms for hMSCs to expand and not undergo spontaneous osteogenic differentiation. Without osteoinduction media, osteogenic markers remained primarily downregulated, consistently so on the NH3-treated 3D printed scaffolds. In 2D, the hMSC population only began to upregulate osteogenic markers at day 22, where this behavior was lacking during the time course in 3D. This suggests that the gained biomimicry from the 3D platform helps to maintain the population. 48 The compliment of 3D geometry and underlying chemistry was sufficient to expand hMSCs without osteogenic differentiation.
With the addition of osteogenic media, hMSCs began to express early osteogenic markers (RUNX2 and ALP). RUNX2 has been theorized as a master regulator, and marker for osteogenic differentiation.49,50 ALP expression during osteogenic differentiation typically peaks, and then expression should decrease as later osteogenic markers begin to be expressed and mineralized matrix is deposited. 51 These early markers typically peak after 1–2 weeks of culture.52,53 With the hMSCs on NH3 treated constructs, earlier peaking expression was observed, consistent with previous results.11,13 The differences in early osteogenic marker expression indicate that hMSCs adhered to O2 and NT substrates may react more slowly to soluble chemical signaling in the media.
As osteogenesis progress, later osteogenic markers, including calcified matrix and OPN, will typically increase with osteogenic differentiation time. 54 This behavior was generally observed under induced osteogenic conditions. Particularly, increasing OPN expression was observed on the NT 3D scaffold under directed differentiation, indicating that the lack of surface functionality could be supporting the later stages of osteoblastic differentiation. Cumulatively, these data show the NH3-treated surfaces and scaffolds supported directed osteogenic differentiation. With osteogenic commitment not solely observed based on surface chemistry, an interaction between the soluble chemistry in the induction media and underlying surface chemistry could be helping to regulate differentiation.
Following directed differentiation, we investigated our scaffold construct under dynamic culture. The chosen flow was expected to encourage proliferation without differentiation, as the estimated shear stress is an order of magnitude below typical values used to induce osteogenesis.32,33 With a flow rate of 3.4 mL/min, we aimed to develop a bioreactor where flow would aid in nutrient and O2 exchange without spontaneous osteogenic differentiation, leading to long-term proliferation of contained hMSCs.
Continued development of this culture system could see the possibility of coculture and using an adherent hMSC population to direct a nonadherent population, as is an approach in hematopoietic stem cell (HSC) expansion. 55 The greatest promise was observed with the NH3-treated scaffolds, maintaining expansion in the bioreactor for at least 7 days. We suggest that the dynamic culture environment has sheared adhered proteins or cells from the NT and O2 scaffolds, where the NH3 treatment aided in retaining these.
The bioreactor system developed with the NH3-treated scaffolds was capable of expanding hMSCs without spontaneous osteogenic differentiation, as demonstrated by the consistent downregulation of early osteogenic markers across treatments. The higher relative calcification measured is likely due to the low number of cells present on the NT surface, as confirmed visually, reducing the total amount of stain available to solubilize and amplifying relative increases. Interestingly, OPN expression increased exponentially on the NT scaffolds inside the bioreactor. This could indicate a link between (a lack of) surface functionality and flow within the dynamic bioreactor environment on this later-stage differentiation marker. In addition to being known as a later-stage osteoblastic marker, OPN is thought to regulate population size in the HSC niche by inhibiting proliferation. 56 Continued investigation into intercellular communication molecules has future use in a targeted bioreactor for the bone marrow niche, with coculture of up to 4 cell populations per line possible.
Conclusions
The goal of this study was to develop a 3D printed, plasma activated PS culture substrate, which encouraged hMSC expansion without inducing osteogenesis. We found that the NH3 treatment is a particularly effective approach for treated 3D printed PS scaffolds and cell systems, supporting hMSC proliferation in 2D, 3D, and 3D dynamic conditions superiorly to NT and O2-treated surfaces and scaffolds. The underlying surface chemistry was insufficient to solely induce differentiation, but supported directed osteogenic differentiation. With osteogenic media, the NH3 scaffolds enhanced early osteogenic differentiation, in agreement with previous literature. PS scaffolds with an NH3 treatment best supports hMSC proliferation and intended osteogenic differentiation. This method results in consistent hMSC expansion on 3D printed PS scaffolds in both static and dynamic culture for up to 7 days within a self-contained bioreactor model. This creates a platform for dynamic culture modeling and studying multicellular interactions found in the bone marrow niche in vitro. Future work seeks to continue developing the system to target coculture interactions within the bone marrow niche.
Footnotes
Acknowledgments
Authors thank Dr. Shin Muramoto and Dr. Greg Gillen of the Surface and Trace Chemical Analysis Group at the National Institute of Standards and Technology for their assistance and fruitful discussions, the Sheikh Zayed Insitute for Pediatric Surgical Innovation for use of their EnvisionTEC 3D Bioplotter, TerrapinWorks for the use of their laser engraver, the Biotherapeutic Development and Delivery Laboratory for the use of incubator space, and Amy Beaven and the University of Maryland's Bioscience Imaging Core for use of their space and equipment. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the Food and Drug Administration, Department of Health and Human Services, or United States Federal Government.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Maryland Stem Cell Research Fund under award no. 2015-MSCRFI1717 (J.P.F.), NIST fellowship program 70NANB15H274 (M.J.L.), and by and the National Institute of Biomedical Imaging and Bioengineering/National Institutes of Health (NIBIB/NIH) Center for Engineering Complex Tissues P41 EB023833 (J.P.F., A.G.M.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.