
Abstract
Tissue-engineered small intestinal implants are being widely investigated as a potential treatment for children with short bowel syndrome, yet are currently limited by their growth potential and relatively low surface area. To address this gap in the field, several investigators have utilized whole organ decellularization of the small intestine as a platform for subsequent growth of intestinal tissue. However, such scaffold-cell constructs require sterilization as a prerequisite for implantation, and the effects of the different pathogen-clearance techniques used on the tissue architecture remains unknown. The effects of four different published protocols for pathogen clearance of decellularized intestine, namely 0.1% peracetic acid (PAA), 0.18% PAA +4.8% ethanol (EtOH), 0.08% PAA +1% hydrogen peroxide (H2O2), and ultraviolet (UV) sterilization were compared using qualitative and quantitative techniques to assess changes to the extracellular matrix, cytocompatibility, and biocompatibility. All methods of sterilization of decellularized intestine were found to be equally effective and each method had similar histologic and scanning electron microscopy appearance of the sterilized tissue. In addition, collagen and glycosaminoglycan quantities, and the ability to support cell growth were similar among all methods. This study provides insights into the change in crypt villous architecture of the extracellular matrix with all sterilization techniques studied. Our findings demonstrate that sterilization affects the microarchitecture significantly, which has not been well accounted for in studies to date, and we were unable to identify a single best agent to achieve tissue sterilization while preserving the microarchitectural features of the tissue.
Impact statement
This work identifies the optimal approach for sterilization of decellularized ileum while preserving tissue architecture. The current findings reveal the ability of the decellularized intestine to support intestinal cell growth in vivo, and reveal the inflammatory response that is induced. Together this work reveals important methodology for the generation of an artificial intestine.
Introduction
Short bowel syndrome is a major cause of morbidity in children, characterized by a lack of intestine leading to inadequate absorption requiring specialized nutritional therapy.1,2 Management of short bowel syndrome includes diet optimization, administration of intravenous nutrition, surgical lengthening procedures, and in the most severe cases, intestinal transplant, which is accompanied by a requirement for lifelong immunosuppression. 3
In view of the ongoing morbidity in patients with short bowel syndrome, several groups including our own4–8 have worked toward the development of an artificial intestine to restore intestinal length, and to offer the potential for enteral autonomy. The use of engineered small intestinal grafts reseeded with the patient's own stem cells would obviate the need for immunosuppression and provide a more sustainable solution for the treatment of intestinal failure and short bowel syndrome. The success of this approach in animal models of intestinal grafts populated by intestinal stem cells have shown promise,4,9–11 and have provided proof of concept for therapeutic use. 9
A few of the major barriers identified in the successful development of an artificial intestine are the length of scaffold required and the lack of appropriate architecture of synthetic scaffolds. Additional barriers which need to be overcome in the development of functional tissue-engineered small intestinal tissue include lack of vasculature and vascular transitions from a macrovascular to a microvascular system, which may be more attainable to achieve with perfusion decellularized intestinal grafts than with synthetic materials.
In an attempt to overcome these barriers, various investigators have examined the role of decellularization of native intestine,12–14 which may be removed from an animal, or potentially even a donor patient. Successful decellularization permits the removal of native cells and most antigenic stimuli, while preserving the tissue architecture and supporting the growth of exogenously plated intestinal stem cells which could be expanded in culture. 15 While techniques for optimal decellularization of intestine have been explored,16,17 techniques for sterilization–which remain critically important for the ultimate clinical utility of an artificial intestine based upon a decellularized matrix—remain incompletely understood. More specifically, the effects of sterilization on decellularized intestinal scaffolds have not been adequately explored.
Previous studies which have evaluated sterilization methods have focused on various techniques ranging from chemicals to ultraviolet (UV) light. One chemical, dilute peracetic acid (PAA) demonstrates potential as a readily available and easy to use option alone or in combination with other oxidation agents. UV sterilization is another attractive option due to its ease of use and short duration to achieve sterilization when compared to other methods. However, a preferred sterilant has yet to be identified for decellularized small intestinal scaffolds. In addition, there have been few reports on how these techniques affect the ultimate structure and function of the scaffolds and thus the potential ability of the decellularized and sterilized scaffolds to support the growth of intestinal stem cells remains unknown.
The goal of this study was to examine previously published protocols for the decellularization and subsequent sterilization of rat small intestine and compare their qualitative and quantitative effects on the decellularized graft as well as the implications for stem cell seeding and reimplantation (described in Fig. 1). In literature review, four methods were identified and replicated as published for evaluation and comparison. Specifically, we evaluated the use of PAA alone18,19 and in combination with dilute ethanol (EtOH) 20 and dilute hydrogen peroxide (H2O2), 21 and nonchemical UV crosslink sterilization. 14 We sought to compare these four methods of sterilization to determine whether there may be differences on tissue architecture, support of intestinal stem cell growth, or inflammatory or infectious response after implantation in vivo. After sterilization, the decellularized tissue was stored in phosphate-buffered saline (PBS) with antibiotic/antimycotic solution replicating the methods reviewed.

Experimental model for the study. The intestine was harvested from adult Lewis rats. The SMA and intestinal lumen were isolated and utilized for perfusion decellularization. The intestine was then sterilized using one of four possible sterilization techniques and the resulting tissue was analyzed to compare the effects of each method of sterilization. SMA, superior mesenteric artery.
Materials and Methods
Reagents, antibodies, and materials
1% antibiotic/antimycotic solution in PBS (1% PBS AA) with 10,000 U/mL penicillin and 0.1 mg/mL of streptomycin (15140122; Gibco) with 0.5 mg/mL amphotericin B (MT30003CF; Corning)
1% (v/v) Triton X-100 (BP151-100; Fisher BioReagents) with 0.1% (v/v) ammonium hydroxide (A669S-500; Fisher Chemical) in dH20
24 g angiocatheter (SR*FF2419; Terumo)
27 g anterior chamber catheter (8065420120; Alcon Laboratories)
4-0 Biosyn suture (USUCM14M; Covidien)
4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI, 422801; BioLegend)
Alcian Blue (A5268-10G; Sigma-Aldrich)
CMGF-media: Cell media growth factor negative (CMGF) consisted of Advanced Dulbecco's modified Eagle's medium (11320-082; Invitrogen), 2 mM Glutamax (35050-061; Invitrogen), 10 mM HEPES buffer (83264; Sigma), 1 × N2 supplement (17502-048; Invitrogen), 1 × B-27 supplement minus Vitamin A (12587-010; Invitrogen), and 1 × primocin (ant-pm-2; Invitrogen) as described22–25
Glycosaminogylcans Assay Kit (6022; Chondrex)
Hydroxyproline Assay Kit (MAK008; Sigma-Aldrich)
Lithium heparin collection tubes (367960; BD Microtainer)
Low Flow Variable Flow Pump (13-876-1; Fisher Scientific)
Mouse C Reactive Protein SimpleStep ELISA Kit (ab222511; Abcam)
Mouse minimal media: CMGF-, 100 ng/mL Wnt3A, 1000 ng/mL R-spondin 1100 ng/mL Noggin, 1 × B-27 supplement, 1 mM N-Acetylcysteine, 50 ng/mL EGF, and 1 × primocin. Y-27632 (Y0503; Sigma-Aldrich)
Papain enzyme (J61875MB; Alfa Aesar)
PicroSirius Red (50-300-77; Electron Microscopy Sciences)
QIAamp DNA Mini Kit (51304; Qiagen)
TrypLE (2605-028; Gibco)
Ultra-Low Variable Flow Pump (13-876-4; Fisher Scientific)
Preparation of decellularized intestine
All surgical procedures were carried out in accordance with Johns Hopkins University Animal Care and Use Committee ACUC Protocols RA18M26 and MO17M304. In brief, a total of 36 adult Lewis rats were euthanized using CO2 inhalation and cervical dislocation. A midline longitudinal laparotomy incision was made to expose the abdominal cavity. A total abdominal colectomy was performed and the colon discarded. The superior mesenteric artery (SMA) of the animal was isolated and cannulated with either a 24 g angiocatheter or a 27 g anterior chamber catheter depending on the size of the SMA. Twenty milliliters of 1% antibiotic/antimycotic solution in PBS (1% PBS AA) with 100 U/mL penicillin and 0.1 mg/mL of streptomycin with 0.5 mg/mL amphotericin B was flushed through the vascular cannula. The cannula was sutured in place. The small intestine was isolated and transected at the point where the SMA crossed over the intestine at the first portion of the jejunum. The superior mesenteric vein was transected and the small intestine was transferred to a Petri dish. The intestinal lumen was intubated with a plastic transfer pipette which was sutured in place. Approximately 2 cm from the tip of the pipette, the plastic was cut so that the transfer pipette fit over the tip of a 60 mL syringe. One hundred twenty milliliters of the 1% PBS AA solution was flushed through the intestinal lumen to remove any fecal material and debris. At this point, a 2 cm segment of terminal ileum was removed from the specimen and was used as “native rat” in the subsequent microscopic and histologic studies of the tissue to provide direct comparison of the tissue from the same animal pre- and post-decellularization and following sterilization. The intestine was transferred to a clean Petri dish and submerged in 1% PBS AA solution until decellularization.
Decellularization procedure
Decellularization was performed at 4°C. The cannula of the SMA of the intestine was attached to an ultralow variable flow pump. The cannula in the intestinal lumen was attached to a low flow variable flow pump. For all reagents, each solution was simultaneously perfused through the SMA via the vascular cannula at 0.78 mL/h and through the intestinal lumen at a rate of 36 mL/h. The solutions were changed at the site of the pump and the rate was kept constant throughout the duration of the experiment. dH20 was continuously perfused through the artery and infused through the lumen for 3 h and the whole intestine was submerged in dH20. Subsequently, 1% (v/v) Triton X-100 with 0.1% (v/v) ammonium hydroxide in dH20 was perfused through the vascular cannula and run through the intestinal lumen overnight. Finally, dH20 was perfused through the artery and circulated through the lumen for an additional 3 h to remove any residual detergent.
Sterilization
The resultant tissue was sterilized using the same vascular and luminal cannulas previously placed for the decellularization process. After the 3 h wash in dH2O step for decellularization, the decellularized intestines were divided into treatment groups. For the first group, the vascular and luminal cannula were perfused with 0.1% PAA for 3 h. For the second group, the vascular and luminal cannula were perfused with 0.18% PAA with 4.8% EtOH for 30 min. For the third group, the vascular and luminal cannula were perfused with 0.08% PAA with 1% H2O2 for 3 h. For the last group, the intestine was transferred to a clean Petri dish, submerged in PBS AA and placed in a UV crosslinker (Stratagene, La Jolla, CA) for one sterilization cycle (90 s) at 254 nm. The dish was covered and one additional sterilization cycle was run. The vascular cannula was flushed with 20 mL of 1% PBS AA and the luminal catheter was flushed with 120 mL of 1% PBS AA. The intestine was submerged in 1% PBS AA for storage at 4°C until use. All scaffolds were used within 2 weeks.
Implantation of decellularized scaffolds into mice
Two centimeters segments of decellularized and sterilized rat ileum were implanted into C57Bl/6 mice under Johns Hopkins University ACUC protocol M017M304. These implants contained decellularized tissue only without seeding the implants. Mice were first anesthetized with isoflurane. The abdominal hair was clipped and skin was prepared using 70% EtOH followed by betadine swab. A midline laparotomy was then performed and the omentum was brought into the field. A single 4-0 vicryl suture was used to secure the decellularized tissue to the omentum. The tissue assumed a flattened shape with the lumen collapsed. In parallel, “surgical control” mice underwent placement of a single vicryl suture without implantation of any decellularized tissue. The abdomen was then closed in two-layers with 4-0 Biosyn suture. All mice received 0.1 mg/kg buprenorphine via subcutaneous injection in the posterior leg following the procedure. “Anesthesia control” mice underwent hair clipping only, and received isoflurane anesthetic, postoperative buprenorphine injection, but no laparotomy. At 7 days, mice were euthanized using isoflurane overdose and cervical dislocation and the omentum with the implanted tissue was harvested for analysis.
Histologic analysis of decellularized tissue
Samples were freshly mounted in tissue freezing medium and cryosectioned at 8 μm. Sectioned samples were fixed on slides with 4% paraformaldehyde in PBS for 1 h (decellularized tissue) or 24 h (native tissue). Tissue sections were stained with hematoxylin and eosin (H&E) (Sigma-Aldrich), Alcian Blue, and PicroSirius Red. Images were taken on EVOS (FL Auto; ThermoFisher Scientific) at 200 × magnification. Images of PicroSirius Red-stained tissue were taken with polarized filters on a Nikon eclipse Ci microscope with a Nikon Digital Sight DS-Fi2 camera.
Enzyme-linked immunosorbent assay of mouse serum
Blood was collected via cardiac puncture just before euthanasia, placed in lithium heparin collection tubes, and centrifuged at 2000 g for 10 min to separate the plasma which was then diluted 1:5000 and run on a Mouse C Reactive Protein SimpleStep ELISA Kit per manufacturer's instructions.
Scanning electron microscopy
Scanning electron microscopy (SEM) was performed according to our previously published protocols. 6 In brief, samples of native intestine collected before the sterilization process and decellularized tissue were fixed in 5% glutaraldehyde in 3 mM MgCl2 and 0.1 M sodium cacodylate buffer, pH 7.2, overnight at 4°C. Samples were rinsed three times for 15 min each in 3 mM MgCl2, 3% sucrose in 0.1 M sodium cacodylate buffer. Following these rinses, specimens were fixed in 1% osmium tetroxide in 3 mM MgCl2 in 0.1 M sodium cacodylate buffer for 1 h on ice. After this fixation, specimens were rinsed twice in deionized water for 5 min each before undergoing dehydration in graded series of EtOH. The final three changes in the dehydration were a 1:1 ratio of 100% EtOH and hexamethyldisilazane and the samples were dried in a dessicator. Once dry, the intestine was incised longitudinally along the antimesenteric border and mounted on stands with carbon tape and then sputter coated with gold-palladium. Samples were subsequently imaged with the Leo 1530S scanning electron microscope (Zeiss, Germany).
DNA quantification
Fresh or decellularized tissue was weighed and homogenized. Approximately 25 g of tissue was obtained from each sample. DNA was isolated using the QIAamp DNA Mini Kit per the manufacturer's instructions. The amount of DNA present in each sample was calculated based on measurements of absorbance at 260 and 280 nm with an Epoch microplate spectrophotometer (BioTek, Winooski, VT).
Collagen quantification
Tissue samples were kept at −80°C until the time the assay was performed. Ten milligrams of tissue was added to a pressure tight polypropylene vial with polytetrafluoro-ethylene-lined cap with 100 μL of deionized water. The Hydroxyproline assay kit was used according to manufacturer instructions. Absorbance was measured at 560 nm using an Epoch Microplate Spectrophotometer (BioTek).
Glycosaminoglycan quantification
Papain digestion of tissue was performed. The following components were mixed: 50 mL of 0.2 sodium phosphate buffer, 400 mg sodium acetate, 200 mg EDTA disodium salt, and 40 mg cysteine HCl. All components were dissolved and then 250 μL of papain suspension containing 5 mg of enzyme was added. Samples were weighed (range 20–53.5 mg) and combined with 1 mL papain extraction reagent and heated in a heating block at 65°C for 3 h. Tubes were centrifuged at 10,000 rcf for 10 min and supernatant was decanted off the top. The glycosaminoglycan (GAG) content was analyzed using the Glycosaminogylcans Assay Kit per the manufacturer's instructions. Absorbance was measured at 520 nm with a SpectraMax M3 Microplate Reader (Molecular Devices, San Jose, CA).
Cell culture
Enteroids were isolated from P7-P15 C57Bl/6 and green fluorescent protein (GFP) expressing C57BL/6-Tg(UBC-GFP)30Scha/mice according to previously published protocols22,24 with some modifications as previously described by our group. 6
Tissue seeding
Following sterilization and storage at 4°C in 1% PBS AA, 3 cm segments of decellularized ileum were isolated. Grossly, the lumen of the intestine was indistinguishable from the serosal side of the intestine following decellularization. To ensure that the luminal side of the tissue was being seeded, the intestine was threaded with a 3.5 cm length of pediatric feeding tube to keep the lumen open. The tissue/tube construct were placed individually in a six-well cell culture plate. A 1.5–2 cm slit was made along the antimesenteric side to expose the lumen leaving both ends intact around the feeding tube. This construct allowed identification of the luminal side at all times grossly. Two hundred fifty microliters of GFP-enteroid cell suspension (cell count 1.1 million −1.5 million cells/mL) in mouse minimal media (passages 5–18) was seeded on the luminal side of the decellularized tissue. The cells were incubated at 37°C for 30 min. An additional 4 mL of mouse minimal media was added to each well to completely submerge the tissue/feeding tube unit. The mouse minimal media was changed every 2–3 days. Analysis of the cells on the decellularized tissue was performed after 7 days of growth.
Immunofluorescent staining of whole-mounted tissue
Whole tissue specimens were fixed in 4% paraformaldehyde at 4°C for 30 min, then rinsed in PBS × 5, the specimens were subsequently counterstained with DAPI as in the protocol described by our group, 7 and evaluated on a Nikon Eclipse Ti confocal microscope (Nikon, Melville, NY). Images were processed using FIJI 26 with the Image 5D plugin.
Live–dead staining and flow cytometry
Decellularized tissue seeded with GFP enteroids were split lengthwise and the feeding tube used to stent open the intestinal lumen was removed. These samples were transferred to a six-well plate. Five milliliters of TrypLE were added to each well for 10 min at 37°C. Five milliliters of CMGF- was added to each well and the plate was gently agitated. The solution surrounding the tissue was pipetted from the plate and transferred to a 50 mL conical tube containing an additional 10 mL of CMGF-. Each remaining decellularized tissue was kept in its respective well and washed with an additional 10 mL of PBS, which was subsequently pipetted from the plate and added to the conical tube to make the total volume of each tube 30 mL. These samples were centrifuged at 750 rcf for 5 min. The pellet was not visible after centrifugation so the supernatant was aspirated leaving ∼3 mL in the bottom of the conical tube. The remaining solution was transferred to a 15 mL conical tube. An additional 5 mL of PBS was used to wash the 50 mL tube and was transferred to the 15 mL tube. Centrifugation was again performed at 750 rcf for 5 min. The supernatant was aspirated. The remaining pellet was suspended in FACS buffer to a total volume of 2 mL. A tube filter was used to filter out any debris, and the sample was transferred to a FACS tube. The resulting cell suspension was diluted 1:10. Samples were stained with 5 μL of 7AAD per tube for 5 min. 7AAD counting was performed on a BD Accuri C6 flow cytometer and analyzed on FlowJo software.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 7.0c (GraphPad Software Incorporated, La Jolla, CA). Normal distributions were assumed. A two-tailed t-test was performed to compare the DNA content between native tissue and decellularized tissue. Analysis of variance (ANOVA, one-way) was used for all other statistical comparisons with Bonferroni-corrected post hoc t-tests if appropriate. ANOVA was performed to compare decellularized intestinal tissue to the sterilization methods only. Native tissue was not included in these comparisons because the control group was the decellularized, unsterilized intestine. Statistical significance was taken as a p-value of less than 0.05.
Photography
All photographs were taken using the camera on iPhone 6S (Apple). Images were edited using Microsoft PowerPoint and ImageJ software.
Results
Decellularization of murine intestine preserves native intestinal architecture
Successful decellularization was accomplished and confirmed histologically and by demonstration that there is minimal DNA remaining in the tissue following the perfusion decellularization process (Fig. 2). Grossly, the appearance of the tissue following the decellularization process was translucent (Fig. 2A, B). Histologic examination of the tissue using H&E shows preservation of the basement membrane and collagen projections consistent with villi into the luminal side of the intestine after decellularization with no nuclear material remaining in either the basement membrane or the luminal components of the tissue (Fig. 2C, D). Examination of the tissue under SEM demonstrated preservation of the villous projections on the luminal side of the intestinal tissue with an organized reticulated pattern in the residual crypt spaces (Fig. 2E, F). DNA quantification was performed on native and decellularized intestinal tissue. The DNA concentration was significantly reduced in the decellularized tissue (0.027 μg/μL) compared to the native tissue (0.178 μg/μL, p = 0.0011). These findings suggest the efficacy of the decellularization process, indicate that tissue architecture is generally well preserved, and lead us to next examine various sterilization techniques.
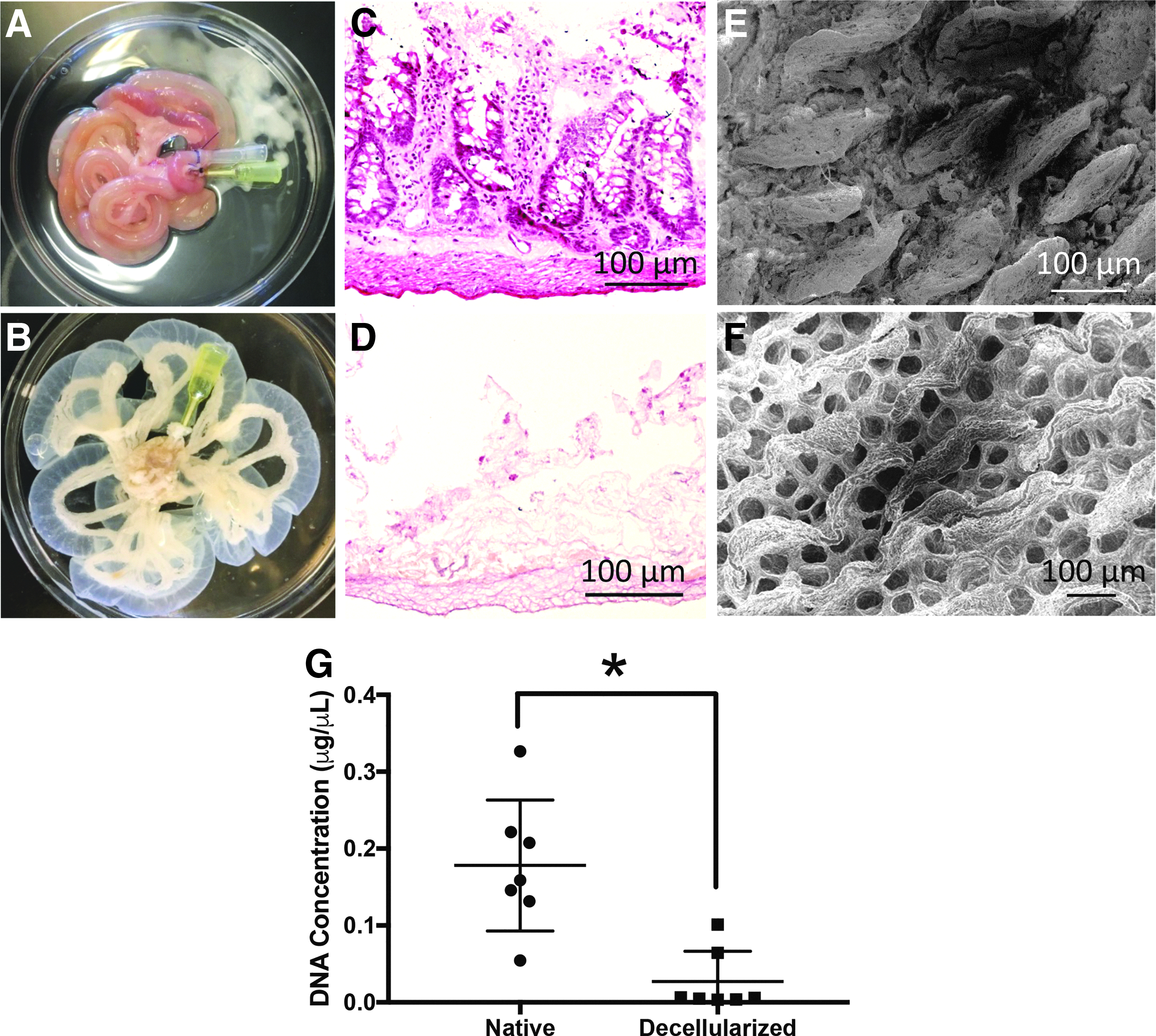
Decellularization of rat intestine.
Histologic comparison of tissue after each of the sterilization techniques
Histologic comparison was performed on each of the groups using several staining techniques to compare the tissue architecture visually (Fig. 3). H&E staining of native tissue demonstrated intact cellular and nuclear material with preserved intestinal crypts and mucin containing goblet cells within the base of these structures (Fig. 3A). Following decellularization of the tissue, loss of cellular structures was observed as well as the loss of nuclear material (Fig. 3B). There were well-preserved extracellular matrix components with debris present at the luminal side. The basement membrane structure was preserved. Chemical sterilization resulted in a similar appearance between each of the treatment groups with a preserved basement membrane and cellular debris retained on the villous side of the tissue. There was little variability observed visually between each treatment using this staining technique.
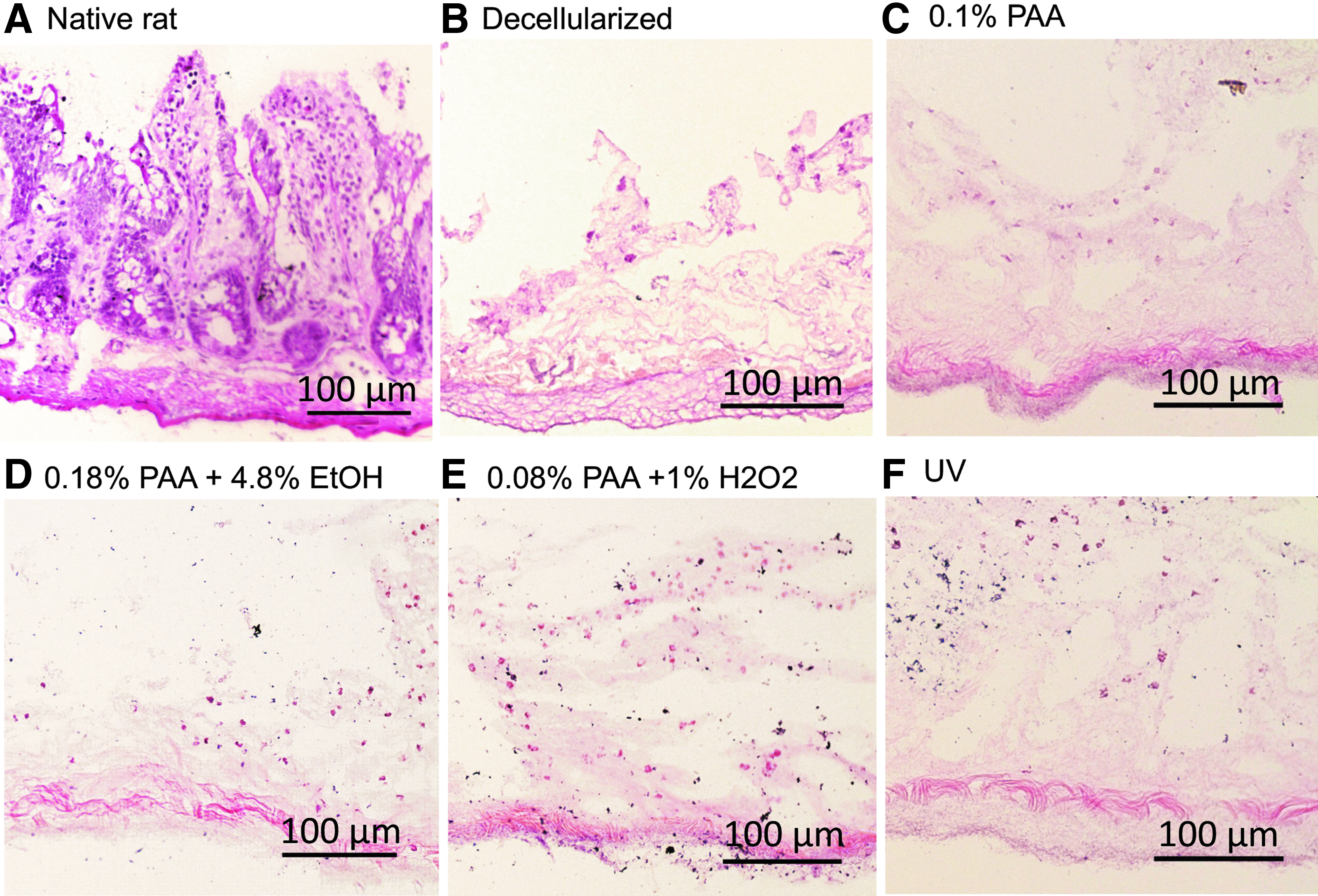
Histologic H&E comparison of native, unsterilized, and sterile decellularized intestine.
Collagen staining of the tissue viewed under polarized light again demonstrated preservation of the collagen structures of the basement membrane. In the native tissue, connective tissue components between the cellular glands are observed in an organized pattern (Fig. 4A). Following perfusion decellularization, these support structures remained relatively unchanged with absence of the previously viewed cellular material (Fig. 4B). The basement membrane remained largely unchanged in appearance with bright birefringence preserved throughout suggestive of a high content of type I collagen. 27 The remaining nonbirefringence is consistent with type IV collagen. 27 Each disinfectant used on the tissue caused a conformational change in the appearance of the mucosal architecture (Fig. 4C–E). The UV sterilized tissue showed relative preservation of the intestinal crypts seen in the native tissue compared with the other treatment groups highlighted by white arrows (Fig. 4F).
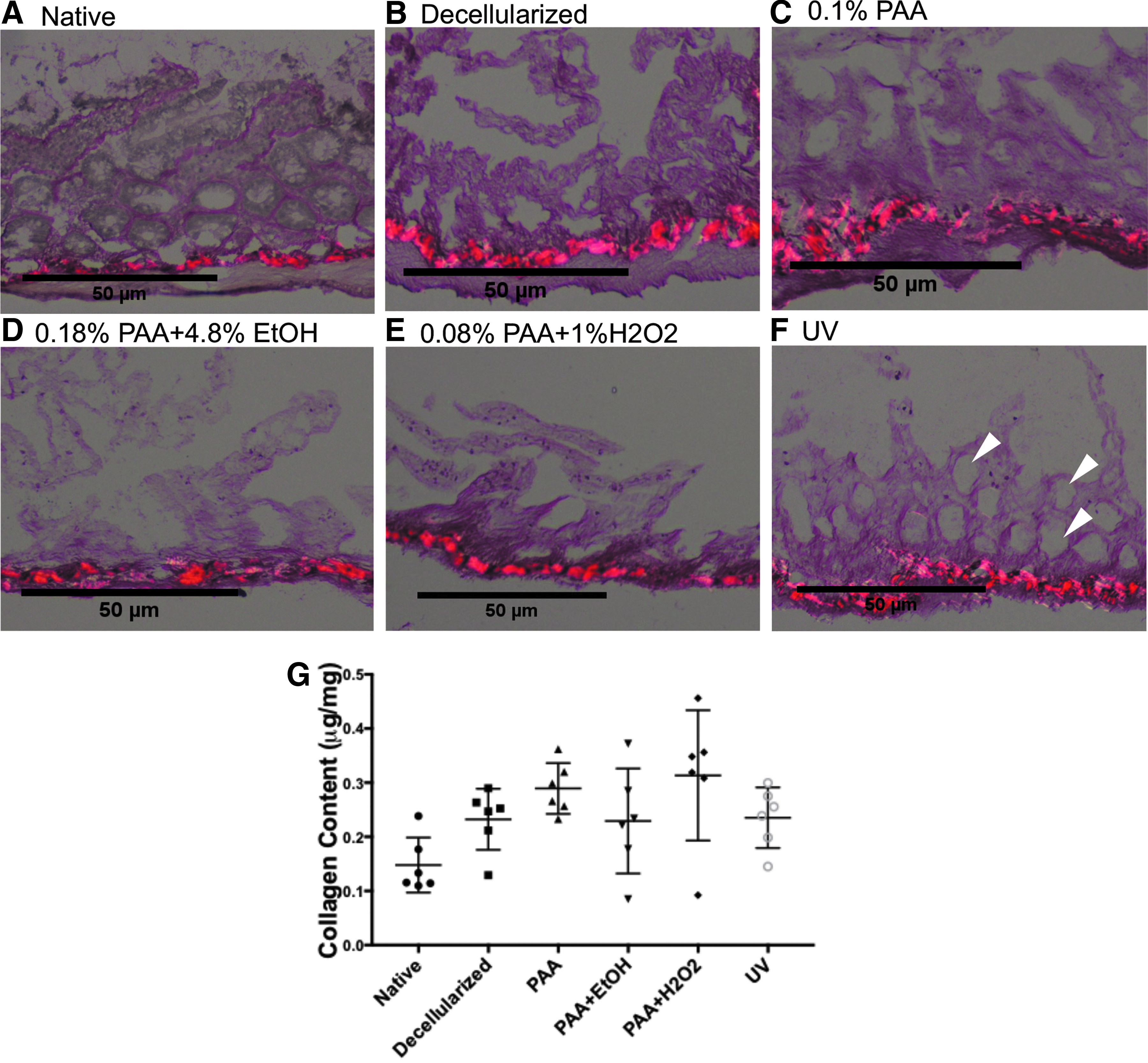
Histologic Sirius red staining of sterile decellularized intestine viewed under polarized light.
Alcian blue staining of the tissues revealed goblet cells of the epithelial layer of native and treated tissues (Fig. 5A). In the decellularized tissue, we observed an absence of strongly positive goblet cells, which was similarly observed in the sterilized tissue (Figs. 5B–F).
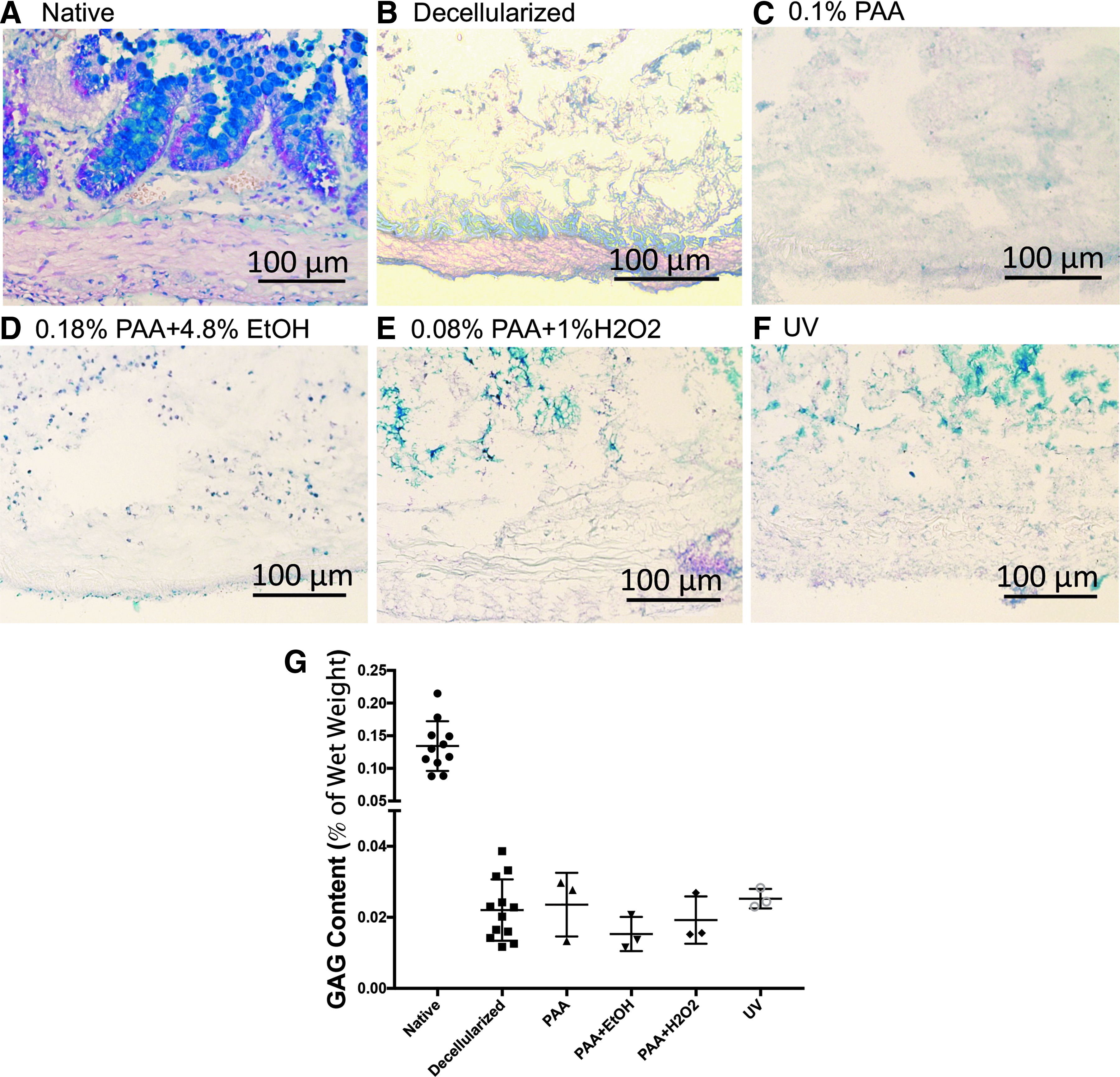
Histologic Alcian blue staining of native, unsterilized, and sterile decellularized intestine at 20 × magnification.
Scanning electron microscopic evaluation of the decellularized intestine after bacterial clearance revealed intact structures
SEM was used to evaluate the changes in the appearance of the tissue both pre- and post-decellularization process and after sterilization (Fig. 6). After perfusion decellularization, the connective tissue that ridges responsible for the structural villous components of the mucosa was well preserved. The crypt portion of the tissue, once decellularized, displayed a regular reticulated appearance between villous projections (Fig. 6B). After sterilization, the crypt portion remained intact along the basement membrane, but additional treatment of the tissue with chemical sterilization caused some loss of tissue architecture with fraying and flattening of the edges at the villous portions of the decellularized tissue scaffold. This change was observed with each of the sterilant solutions studied (Fig. 6C–E). The tissue sterilized with UV light demonstrated a similar flattening of the trabecular regions with preservation of the reticular glandular crypts (Fig. 6F).

SEM comparison of native, unsterilized, and sterile decellularized intestine.
GAG quantification and collagen quantification
Extracellular matrix (ECM) components of native rat intestine, decellularized rat intestine, and decellularized tissue exposed to the four sterilization techniques were evaluated and compared. The GAG content was assessed as a measure of % of wet weight (Fig. 5G). The decellularized tissue had a reduction in GAG content by 83.6% compared to the native rat tissue (0.022% ± 0.009%). This percentage of wet weight of GAGs was preserved across treatment groups with no statistical difference in the GAG content of native decellularized rat tissue versus sterilized decellularized tissue between all treatment groups (Fig. 5G, PAA 0.024 ± 0.009, PAA+EtOH 0.015 ± 0.005, PAA + H2O2 0.019 ± 0.007, and UV 0.025 ± 0.003). Collagen quantification was performed with preservation of the overall collagen content of the tissue. There was no significant difference observed between the native tissue, the decellularized tissue, and the sterilized treatment groups (Fig. 5G).
Enteroid cell growth
Enteroids were successfully seeded onto decellularized intestinal tissue, and growth of GFP enteroids on the decellularized scaffold was confirmed on both SEM and confocal microscopy. Once the cells were seeded and grown in culture for 1 week, a comparison of SEM and confocal cell imaging with counterstaining demonstrated that the scaffolds accommodated the cells and facilitated a monolayer formation along the surface of the tissue (Fig. 7A, B). With each sterilization technique tested, seeding of the tissue resulted in cell viability at 1 week with similar numbers of live cells harvested from the tissue following trypsinization, 7AAD staining, and flow cytometry (Fig. 7C). PAA+EtOH had statistically significantly more cells compared with the other groups (PAA+EtOH 21,743 ± 14,894 cells vs. PAA + H2O2 5145 ± 3118 cells with p-value 0.0089; PAA 4917 ± 3445 cells p-value 0.0079; and UV 1273 ± 711 cells p-value 0.0012). This seems to have been driven primarily by two samples in the PAA+EtOH group, but it is possible that this combination is more suitable for cell survival after seeding than the other sterilization techniques. Further confirmation studies will be needed.
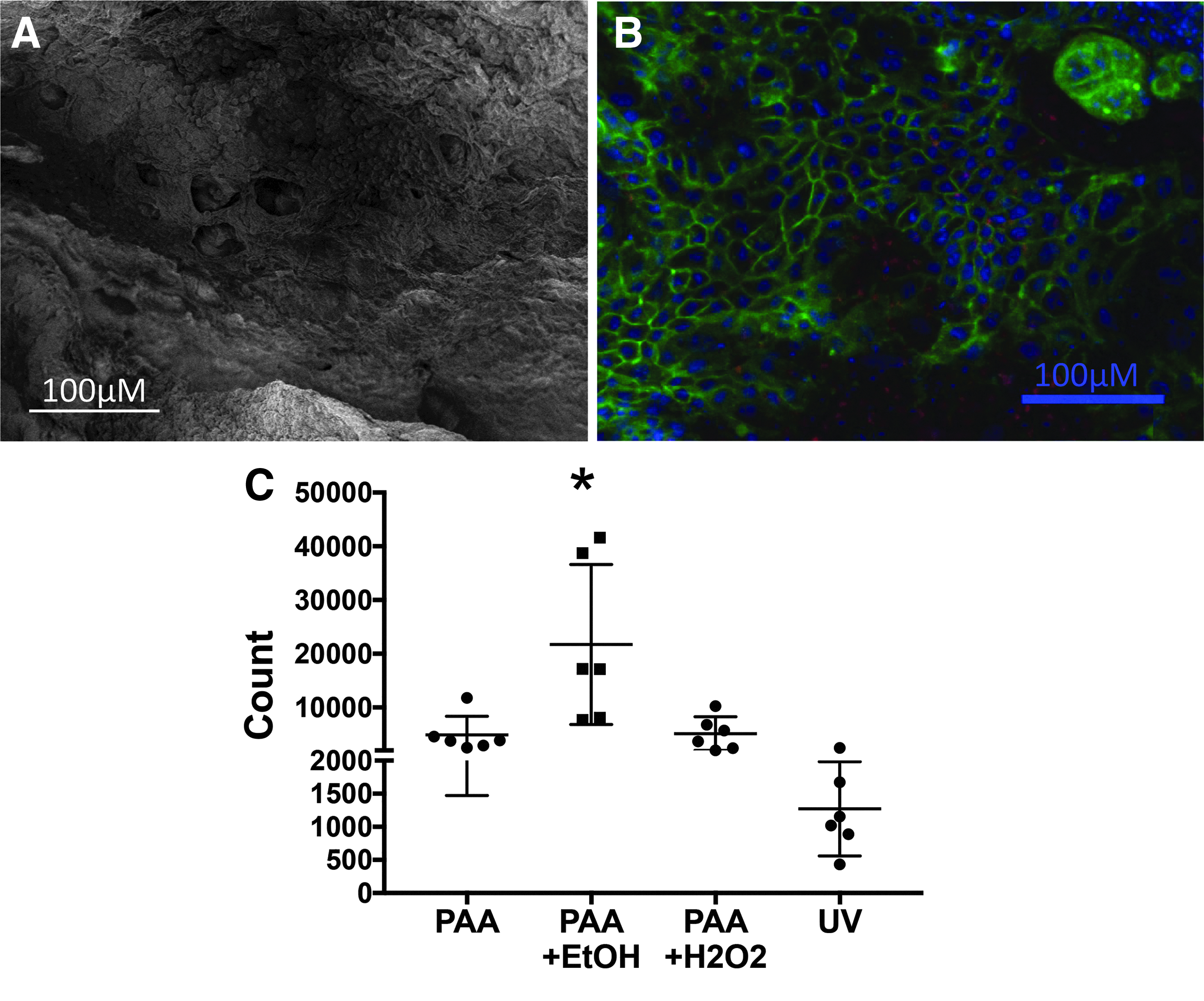
A comparison of SEM
In vivo inflammatory response
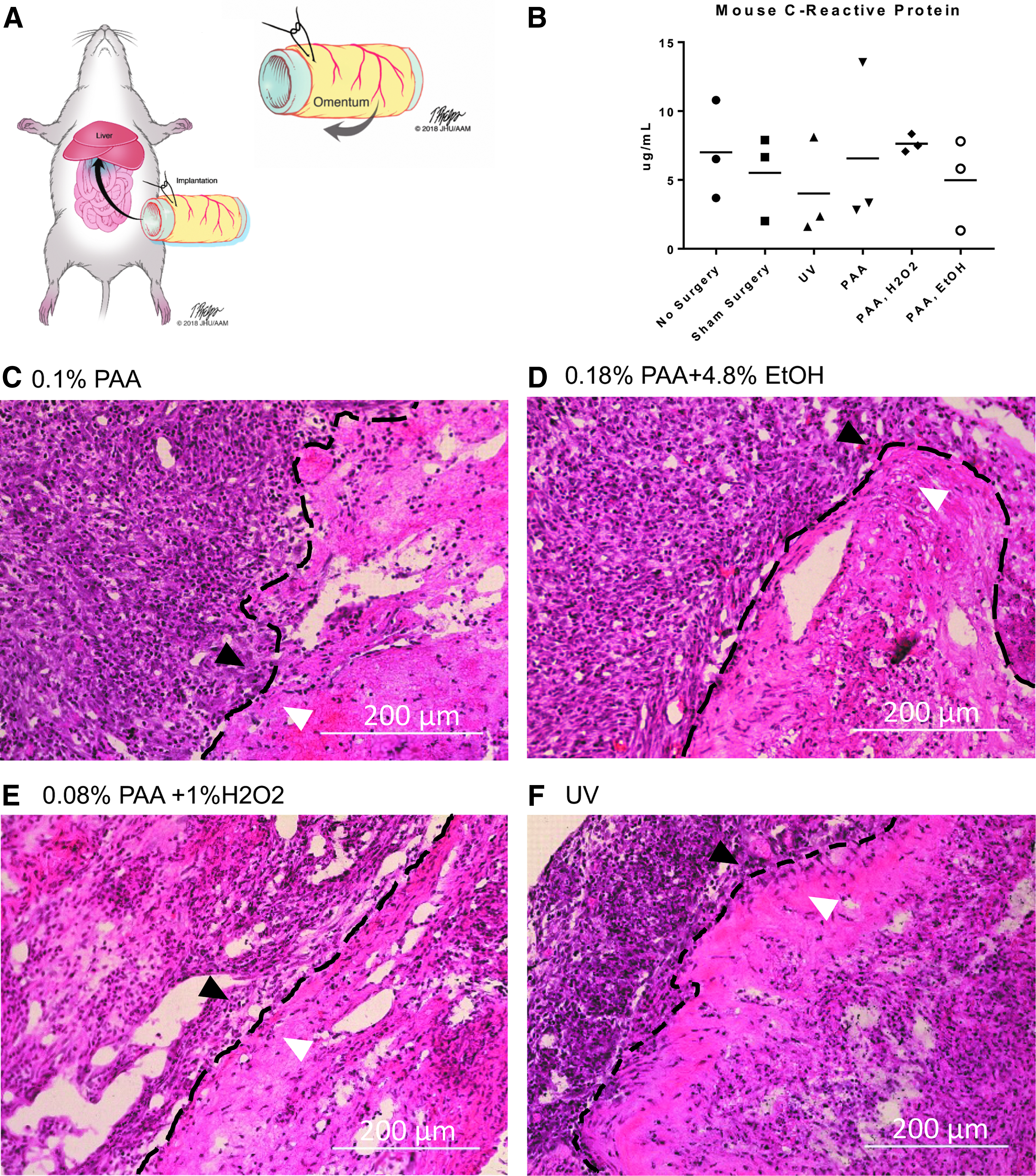
The ability of the decellularized and sterilized tissue to support cell growth in vivo was assessed by implanting the graft in the omentum of C57Bl/6 mice (Fig. 8A, B). At 7 days, there was 100% survival of all mice with implanted tissue (n = 19). C-reactive protein enzyme-linked immunosorbent assay (ELISA) was performed on all samples with no significant difference in this inflammatory marker observed between treatment groups (Fig. 8B). Histologic examination of the tissue demonstrated a clear demarcation between the implanted tissue and the surrounding omental tissue with robust inflammatory reaction (Fig. 8C–F).
Discussion
This study demonstrated our ability to perfusion decellularize small intestine using a modified protocol of Dew et al. 19 and to evaluate the effects of four sterilization techniques on the ultimate structure and function of the scaffolds. The intact nature of the intestinal scaffold was confirmed by gross appearance, histologic evaluation, collagen staining, and SEM and was consistent with prior reports.14,19,28
The SEM analysis showed the three dimensional changes in tissue architecture with sterilization. Across all chemical treatment groups as well as the UV sterilized tissue, the villous projections of ECM were blunted compared to the decellularized tissue. This suggests that any of the sterilization protocols followed results in a partial loss of the villous architecture. This is similarly observed with the loss of mucosal architecture seen on histologic examination and discussed. Therefore, sterilization using any of the methods presented may create architectural distortion of the ECM, which has unknown effects on the differentiation of cells seeded on these scaffolds. This architectural loss is problematic as preservation of the ECM of the villus-crypt unit has been postulated to establish the regenerating unit during intestinal cell turnover. 28 If the tissue architecture is not preserved, some of the benefits of using a decellularized scaffold for tissue regeneration are undermined by the chemical sterilization process and a different method of bacterial clearance or sterilization should be sought to preserve these components.
Histologic analysis of the tissue was performed using a frozen section technique rather than paraffin blocks. Some limitations are introduced in the histologic evaluation of decellularized samples. The fragile nature of the tissue following perfusion decellularization caused rapid breakdown of the samples when using EtOH-based fixation and processing techniques. This resulted in the samples crumbling when attempting to cut the paraffin-embedded blocks. The tissue was more tolerant of the tissue freezing medium and cryosectioning, and we observed more preserved tissue integrity and architecture using this technique to section the tissue.
When evaluating the GAG content of native or decellularized tissue, the histologic comparison of the tissues when stained with Alcian blue demonstrates a remarkable loss of strongly acidic mucosubstances, which results in the blue appearance of the tissue. The decellularized tissue and sterilized tissue alike only weakly stain using this protocol. The quantification of this change performed using a standardized kit resulted in an order of magnitude drop of GAG content between the native and decellularized tissue. In the article by Dew et al., which was used for the decellularization protocol in this article, a different technique utilizing lyophilized tissue samples was used to generate GAG quantification in ng/mg of dry tissue. The findings in that article showed a preserved quantity of GAGs and mucopolysaccharides. Other groups 12 have demonstrated a substantial loss in sGAGs during the decellularization process compared to the native tissue. This was the trend observed in our study both histologically and quantitatively with a standard kit GAG assay measuring GAG content in % of wet weight. The lack of observed difference between the sterilization treatments may not be a meaningful finding as the drop in GAG content was observed during the decellularization protocol rather than during sterilization.
Monolayers of intestinal enteroids were able to be grown on decellularized tissue sterilized with each of the methods presented. One observation is the difference in cell growth between the tissue treated with 0.18% PAA +4.8% EtOH and the other groups. While not statistically significant, an observed difference of 10 times the numbers of live cells were harvested from seeded scaffolds on subsequent flow cytometry. As suggested before, additional studies on how the sterilant may impact cell adhesion specific to intestinal ECM need to be performed before suggesting that this may have some advantage over the other methods studied. Also observed was a much lower average harvest of live cells from the UV-sterilized tissue, which also did not reach statistical significance. This study demonstrates biocompatibility of the sterilized grafts with enteroid monolayer growth. SEM evaluation of the tissue architecture pointed out loss of villous ECM projections and we commented that this may impact differentiation of cells dependent on ECM signaling. While this remains a question, sterilization of the scaffolds did not impact epithelial stem cell monolayer formation. The cell seeding methodology for this study was limited to seeding the epithelial surface. Significant improvements in this methodology toward tissue generation have already been demonstrated by other groups in this field using bioreactor systems for culture of multiple cell lineages. 12 As different stem cell lineages are added as necessary components toward a functional whole organ graft, limitations imposed by the chemical reagents studied may be identified.
In vivo, the effect of each of these sterilization methods resulted in similar acute phase immunogenic response to sterilized tissue implantation with 100% recipient survival at 1 week after implantation. This finding supports the efficacy of each method in sterilization of the tissue graft. Clearance of the acute phase reactant c-reactive protein at similar levels across treatment groups at 1 week is comparable to levels of c-reactive protein in control and sham groups, which also support the efficacy of sterilization. The histological appearance of the tissue harvested at 1 week uniformly illustrates lymphocytic infiltration of the implanted graft. Longer time points to verify resolution of the acute inflammation were not examined in this particular study, which raises the question of whether chronic inflammation is an issue when the decellularized tissue is implanted.
The sterilization process of decellularized whole tissue grafts has been valued and studied for other applications, including dermal allografts, 29 tendon, 30 and cardiovascular scaffolds, 31 but before this work, no other groups have tried to identify an optimal sterilization method for decellularized intestinal tissue that preserves the properties of the graft that makes it an attractive scaffold for engineering of the small intestine. This study examined a relatively small field of methods for sterilization in very limited concentrations. One method recently cited as an option for the sterilization of biomaterials is supercritical CO2 sterilization. When applied to the sterilization of a decellularized aortic valve, one group demonstrated successful sterilization of the decellularized with no ECM crosslinking observed as a result of the sterilization process. 32 This may provide promise as a modality for sterilization of the decellularized small intestine that better preserves the microarchitecture. A potential limitation of using this technique on intestinal tissue is the increased pressure required to create the supercritical phase—the fragile nature of the decellularized intestinal tissue may result in delamination as the collagen profile of the intestine is significantly lower in density than valvular tissue.
While our group aimed to elucidate some of the effects of sterilization on perfusion decellularized small intestine, a number of limitations should be mentioned to complete the discussion surrounding this work. This study evaluated only the methods of sterilization for decellularized small intestine that have been previously published specifically for this type of decellularized tissue. As these methods were utilized by other authors in recent publications, verification of bacterial clearance was not performed as a part of this analysis. The success of sterilization for each method was not validated as the goal of this evaluation was to compare the effects of each process on the resulting tissue. This raises the question of sterilization of the tissue in a separate process versus utilization of antibiotics and antifungal agents to accomplish the same goal of pathogen eradication from the tissue scaffold before seeding with new cell populations. One clear barrier to the interpretation of the in vivo implantation of the decellularized rat tissue into the abdomen of a mouse is the cross reactivity that may arise when cross species implants are utilized. As mentioned previously, the protocol selected for the examination of GAG content in the tissue utilized a wet-weight analysis with a standard kit. This type of analysis has been successfully used by other groups 12 to examine the changes that result in GAG content following the decellularization process. A more frequently cited technique for this evaluation involves examination of lyophilized tissue samples to generate GAG quantification in ng/mg of dry tissue.14,19 This method may have provided different outcomes and the two processes should be examined in a side by side comparison to validate which provides a more accurate method of GAG content analysis for this type of tissue. While the decellularization process should eliminate or at least significantly decrease the immunogenic cross reactivity between species, the extent to which this impacts the inflammatory response that was examined in this study remains unknown. Implanting the decellularized tissue in rats would have helped to clarify the extent of inflammatory changes related to the process of implantation. For the interpretation of our data, the observation that all sterilization techniques produced a similar extent of inflammation in vivo is the only observation that can truly be made using our methodology. The cell-seeding methodology utilized in this study was limited surface seeding of the epithelial surface of the decellularized small intestinal tissue. There have been several other groups that have had better success using other methods of cell seeding such as spray delivery of organoids 21 and perfusion of multiple cell lineages in a bioreactor model.12,19 Compared to other methods, our study was limited to the effects of the sterilization process on the ability to general monolayers of a single epithelial intestinal stem cell line. The effects of these processes on tissue and whole organ regeneration remain unknown and a more advanced model for tissue generation would provide a more complete understanding of these effects rather than relying on extrapolation of data from monolayer culture of a single cell line.
As we look toward the introduction of decellularized intestinal grafts in therapeutic trials, understanding each component phase of graft preparation is essential. While intestinal transplant outcomes are improving, a different solution that obviates the need for immunosuppression is needed to improve outcomes for children with short bowel syndrome and intestinal failure. Patient specific intestinal grafts represent a potential therapeutic option to meet these needs and continued investigation is needed to achieve functional tissue-engineered intestinal grafts for therapeutic use.
Conclusions
All methods of sterilization of decellularized intestine were found to be equally effective and each method had similar histologic and SEM appearance of the sterilized tissue. In addition, collagen and GAG quantities and the ability to support cell growth were similar among all methods. This study provides insights into the change in crypt villous architecture of the ECM with all sterilization techniques studied and provides a rationale for additional investigation of techniques that preserve architectural features unique to the small intestinal epithelium. Preserving these microarchitectural features may impact the organization and differentiation of cells seeded on these scaffolds and may impact the function of the regenerating unit. Our findings demonstrate that sterilization affects the microarchitecture significantly, which has not been well accounted for in studies to date, and we were unable to identify a single best agent to achieve tissue sterilization while preserving the microarchitectural features of the tissue.
Footnotes
Acknowledgments
The Hackam group thank Barbara Smith of the JHSOM microscopy core for her assistance with SEM imaging. Additional members of the Hackam Laboratory are acknowledged, including Hongpeng Jia, Peng Lu, Thomas Prindle, Emilyn Banfield, Quinjie Zhou, Jungeun Sun, Diego Nino, Sanxia Wang, and Yukihiro Yamaguchi. The team also thank Rohan Panaparambil for his assistance with enteroid culture. Finally, the high-quality illustrations were drawn by Timothy Phelps of the medical illustration department of the JHSOM.
Disclosure Statement
None of the authors has any commercial association with any entity which could create a conflict of interest in association with the current article.
Funding Information
M.R.L. received salary support for this study under a National Institutes of Health and National Institute of Diabetes and Digestive and Kidney Diseases T32 training grant (2T32DK0077-13-21). A.D.W. received salary support under a National Institutes of Health T32 salary grant (ST320D011089-42).