
Abstract
Objective:
To evaluate a vitrification protocol from histology to gene expression to slow freezing.
Methods:
Ovaries from 12 prepubertal ewes. The same ovary was cut into fragments, studied fresh, frozen, and vitrified. Follicle morphology by hematoxylin-eosin-safran staining, vitality by Trypan Blue, and apoptosis by marking cleaved caspase-3 were studied. The expression of gene: anti-Müllerian hormone (AMH), cytochrome p450 family 11 subfamily A member 1 (CYP11A), and steroidogenic acute regulatory protein (STAR; granulosa cells); growth differentiation factor 9 (GDF9) and zona pellucida glycoprotein 3 (ZP3; oocytes); and cyclin D2 (CCND2) and cyclin-dependent kinase inhibitor 1A (CDKN1A; cell cycle regulation), was evaluated by reverse transcription quantitative polymerase chain reaction.
Results:
The slow freezing protocol had a significant negative impact on intact primordial follicles compared with fresh tissue (37.6% vs. 62.5%, p = 0.003). More intact follicles after vitrification were observed compared with slow freezing (p = 0.037). The apoptotic primordial follicles were similar after slow freezing and vitrification (12.6% vs. 13.9%). Concerning granulosa cell genes, slow freezing led to a trend toward overexpression of AMH messenger RNA (mRNA; p = 0.07); while vitrification led to a significant overexpression of CYP11A mRNA (p = 0.003), and a trend toward an overexpression of STAR mRNA (p = 0.06). Concerning oocyte genes, both techniques did not lead to a difference of GDF9 and ZP3 mRNA. Concerning cell cycle genes, slow freezing led to a significant underexpression of CCND2 (p = 0.04); while vitrification did not lead to a difference for CCND2 and CDKN1A mRNA.
Conclusion:
Vitrification preserved follicular morphology better than slow freezing and led to gene overexpressed, while slow freezing led to gene underexpressed.
Impact statement
The preservation of female fertility and in particular the cryopreservation of ovarian tissue (OT) is a major public health issue aimed at improving the quality of life of patients after gonadotoxic treatments. The use of slow freezing of this OT, which is the reference technique, is not optimal due to tissue alteration. The alternative would be vitrification. This study compares these two techniques. We have highlighted that vitrification preserved follicular morphology better than slow freezing and led to gene overexpressed, while slow freezing led to gene underexpressed.
Introduction
Over the past few decades progress in oncology treatment and diagnosis has improved the health of young women suffering from cancer. However, these cancer treatments (chemotherapy, radiotherapy, and surgery) can induce great damage to ovarian reserve. 1 There are several methods to preserve fertility, including oocyte, embryo, and ovarian tissue (OT) cryopreservation; in prepubertal girls the most promising method is the cryopreservation of OT. Slow freezing is the reference method for cryopreservation of human OT, although it can reduce the ovarian follicular reserve and induce damage to stromal cells. 2
Vitrification is an alternative method that is currently being investigated but has yet to be implemented in routine practice. The aim is to avoid crystal formation to reduce cellular damage, and it seems to be better than slow freezing for the preservation of both the cells and stromal matrix, but also the survival of a vast pool of viable quiescent follicles that represent the ovarian reserve of the graft.3–9 However, authors have reported the superiority of slow freezing compared with vitrification10–14 or no significant difference between the two cryopreservation techniques.15–22 These conflicting results are likely to be due to the various methods employed to vitrify OT and to the various evaluation criteria used (follicular morphology, follicular vitality, apoptosis, and gene expression). Authors studied one or more of these parameters but none considered all of them in the same study.
To the best of our knowledge, there is no published study that has compared the effects of slow freezing with vitrification on OT on gene expression of granulosa cells, oocytes, and cellular cycle. Given the heterogeneity of the evaluation methods, it is therefore necessary to carry out a study with a more complete approach, in particular, including the investigation of gene expression.
The aim of the present study was to evaluate a vitrification protocol from histology to gene expression in relation to the reference technique, slow freezing.
Materials and Methods
The study protocol was approved by the local Ethics Committee (Comité d'éthique des Hospices Civils de Lyon) and the veterinarian authority responsible.
Collection of ovaries
Ovaries (n = 12) were recovered from prepubertal ovines (6–8 months of age) at a slaughterhouse of Lyon (Corbas, France). Ovaries were immersed in Leibovitz L-15® (Eurobio, Courtaboeuf, France) and transported to the laboratory at 10°C within 40 min. First, ovaries were cut in two hemi-ovaries using a scalpel in a Petri dish under sterile conditions. Then, the medulla was removed with a sterile chisel to obtain two hemicortexes. Finally, cortexes were cut into small pieces of 150 ± 20 mg representing ∼5 mm (length) × 3 mm (width) × 1 mm (thickness). The same ovary was cut into several fragments to be studied fresh (control tissue), after slow freezing and after vitrification.
Slow freezing protocol and thawing procedure
OT was frozen according to the method described by our team that allowed live birth ewes,23,24 which we have modified by 10% replacing fetal calf serum (Sigma-Aldrich, St Quentin Fallavier, France) with 10% serum substitute supplement (SSS; Irvine scientific, Santa Ana). After OT dissection, fragments were incubated in BM1 (Eurobio) and placed in 800 μL of freezing solution composed of 14.2% (2M) dimethyl sulfoxide (DMSO; Sigma-Aldrich), 10% SSS and BM1 within a sterile straw (CryoBioSystem, L'Aigle, France) for 10 min at room temperature between 20°C and 23°C, then subsequently frozen with a semiautomatic self-seeding programmable freezer (Minicool 40 PC; Air Liquide Santé, France) held at 20°C. The cooling rate was −2°C/min from 20°C to −35°C, at which point temperature nucleation was induced by semiautomatic seeding. The semiautomatic seeding was performed by the release of negative calories at −11°C. Then the temperature was lowered to −150°C at 25°C/min. When the temperature reached −150°C, the straws were plunged in liquid nitrogen for storage.
For thawing, straws were removed and immersed in a water bath at 37°C for 1 min. OT was removed from the straw to three successive thawing solutions composed of BM1 for 5 min at room temperature. Thawed OT was placed into 4% formaldehyde (VWR, Strasbourg, France) for investigation of morphology and apoptosis, and into BM1 for the investigation of follicle vitality.
Vitrification protocol and warming procedure
OT was vitrified in a solution composed of DMSO, ethylene glycol (Sigma-Aldrich), SSS, and sucrose (Dutscher, Brumath, France). The protocol was based on several vitrification protocols described in the literature.17,25–27 Fragments were incubated in equilibration solution (BM1 containing 5.58% [1 M] ethylene glycol, 3.55% [0.5 M] DMSO, 2.50% SSS, and 0.125 M sucrose) for 5 min at room temperature between 20°C and 23°C. Then they were placed into a second vitrification bath (BM1 containing 11.16% [2 M] ethylene glycol, 7.10% [1 M] DMSO, 5.00% SSS, and 0.25 M sucrose) for 7 min at room temperature between 20°C and 23°C, after which they were placed into the vitrification solution (BM1 containing 22.32% [4 M] ethylene glycol, 14.20% [2 M] DMSO, 10% SSS, and 0.5 M sucrose) at 4°C for 10 min.
OT was placed on a piece of semirigid absorbent paper 1 mm thick developed by our team, then was cooled by direct contact with liquid nitrogen and then stored in vials (CryoElite 5 mL; Wheaton, Millville) without contact liquid nitrogen. For warming, OT was removed from the vial and placed in a warming solution (BM1, 0.8 M sucrose) for 5 min at 37°C, then transferred into a solution of 0.4 M sucrose for 5 min at room temperature, and then into sucrose-free BM1 for 5 min at room temperature. Warmed OT was placed into 4% formaldehyde for investigation of morphology and apoptosis, and into BM1 for the investigation of follicle vitality.
Histological evaluation
Twelve fresh, 12 frozen/thawed, and 12 vitrified/warmed OT was fixed in formaldehyde 4% for 24 h at room temperature, paraffin embedded after dehydration, and cut into 4 μm serial sections. Ten sections were stained with Hematoxylin (Millipore, Burlington), Eosin (Sigma-Aldrich), and Safranin (RAL diagnostics, Martillac, France). 28 The entire section was photographed to make a count and a classification of the follicles using the ImageJ software. Then sections were checked by light microscopy to evaluate follicle morphology. The follicles were counted by two blind operators and were classified according to the description of reported by Gougeon and counted as altered (Fig. 1) or intact (Fig. 2). 29 Follicles were classified as altered if there were at least one sign of oocyte or granulosa cell degeneration: the presence of pyknotic oocyte or follicular cell nuclei, detachment of the oocyte from surrounding granulosa cells, vacuolization of ooplasm, partially degenerated granulosa cells, or detachment of the basal membrane.
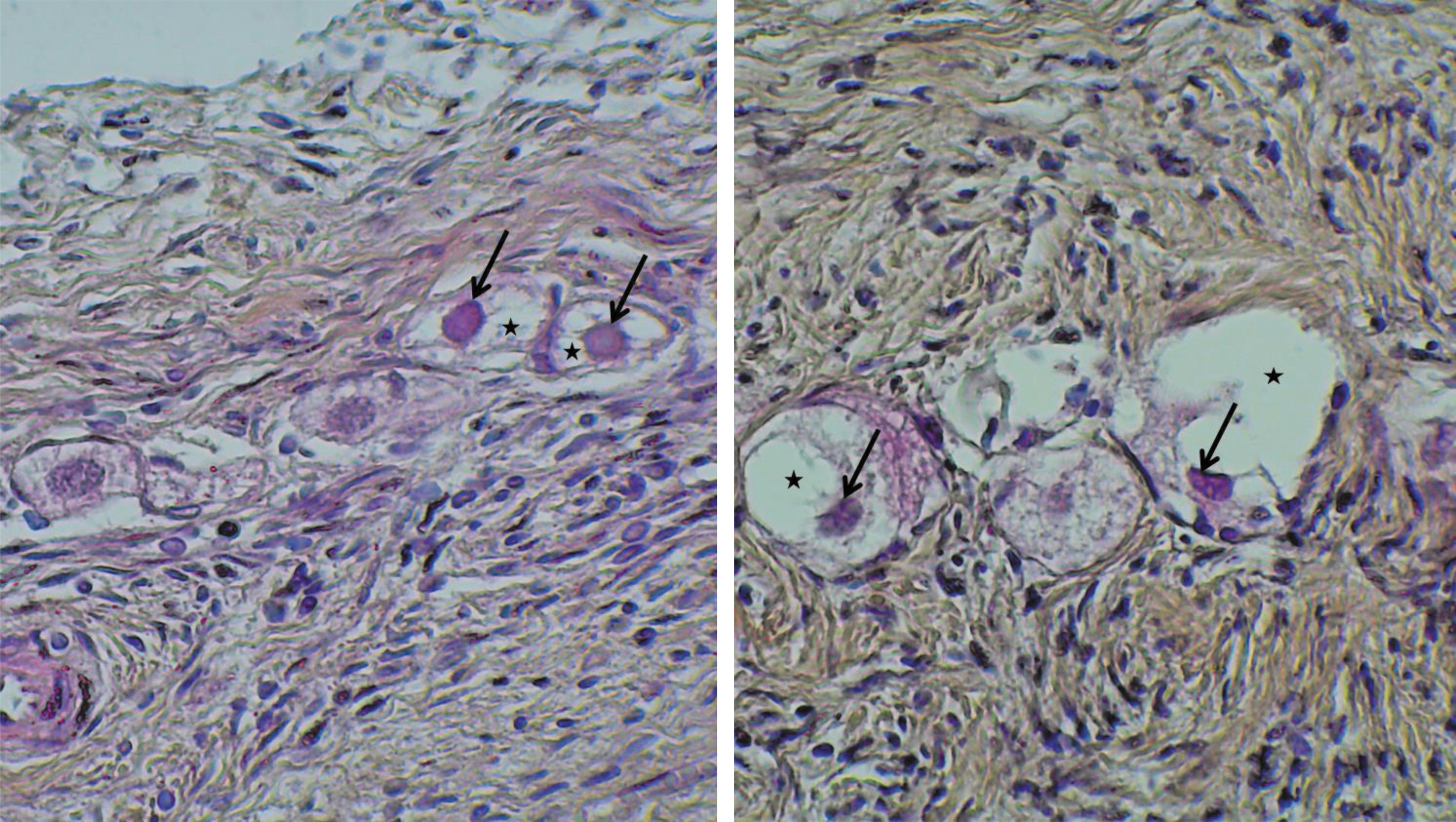
Sections of HES-stained OT at magnification 40. The black arrows show oocyte pyknotic nuclei. The black stars show follicle cytoplasmic detachments. HES, hematoxylin-eosin-safran; OT, ovarian tissue. Color images are available online.
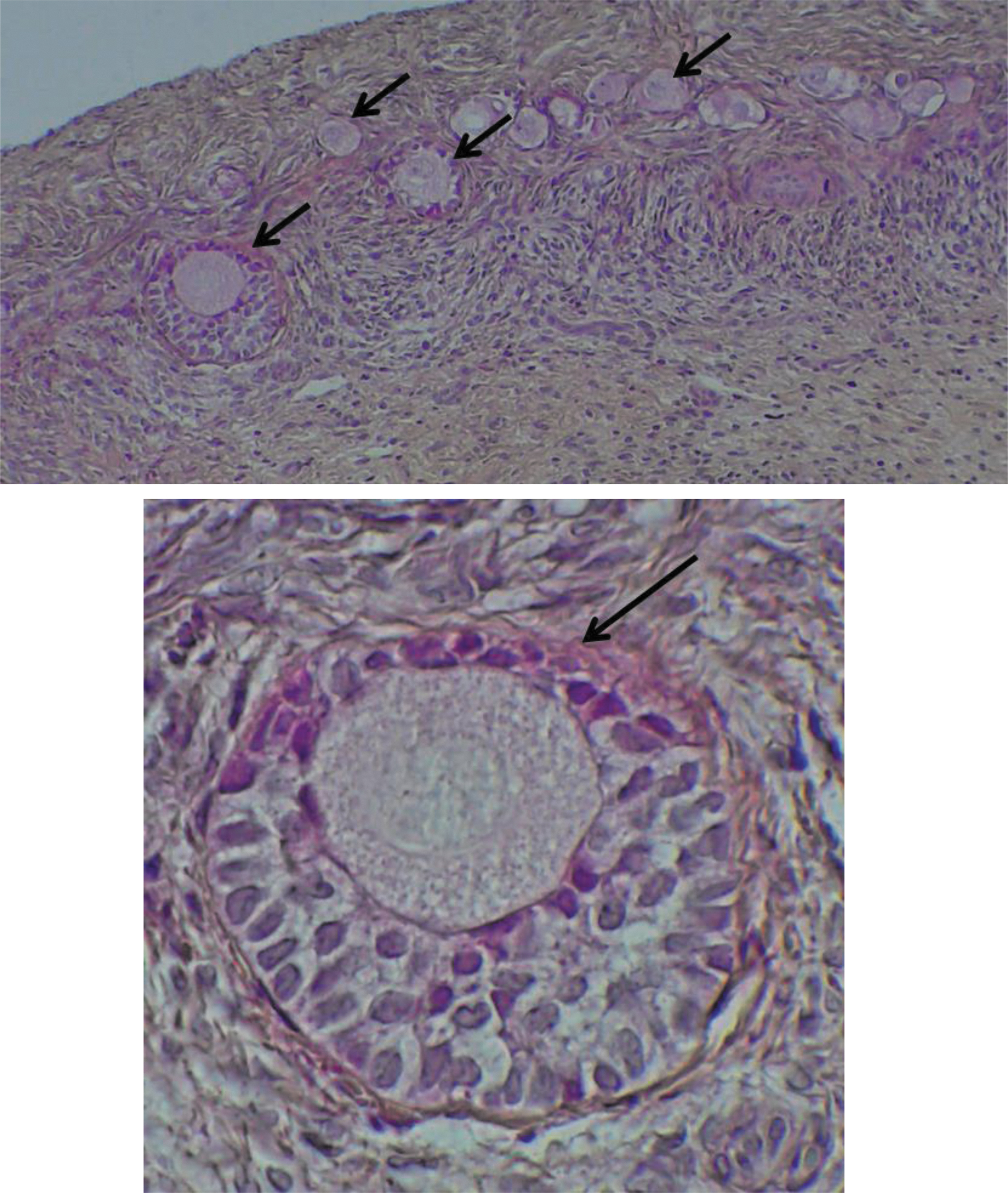
Sections of HES-stained OT at magnification 10 and 40. The black arrows show intact follicles. Color images are available online.
Evaluation of vitality
Fresh, frozen/thawed, and vitrified/warmed OT fragments were cut mechanically into 1 mm2 pieces using a scalpel in a Petri dish and then incubated in a collagenase I solution (Sigma-Aldrich) 23 at 7275 U/mL in a 15-mL Falcon® tube (Corning Life Sciences, Amsterdam, The Netherlands) for 90 min at 37°C in a water bath under regular agitation. After digestion, the enzymatic reaction is stopped by adding phosphate-buffered saline (PBS; bioMérieux, Marcy l'étoile, France) and BSA 0.3% (Sigma-Aldrich) volume to volume. The solution is centrifuged at 363 g during 5 min at room temperature between 20°C and 23°C and the cells were incubated with Trypan Blue (Sigma-Aldrich) and observed by light microscopy to evaluate follicle vitality. All follicles from the fresh, frozen/thawed, and vitrified/warmed fragments were counted and classified into altered or unaltered follicles according to the vital coloration. Follicles in which the oocyte and/or ≥50% of the granulosa cells were stained blue were considered as altered (Fig. 3).
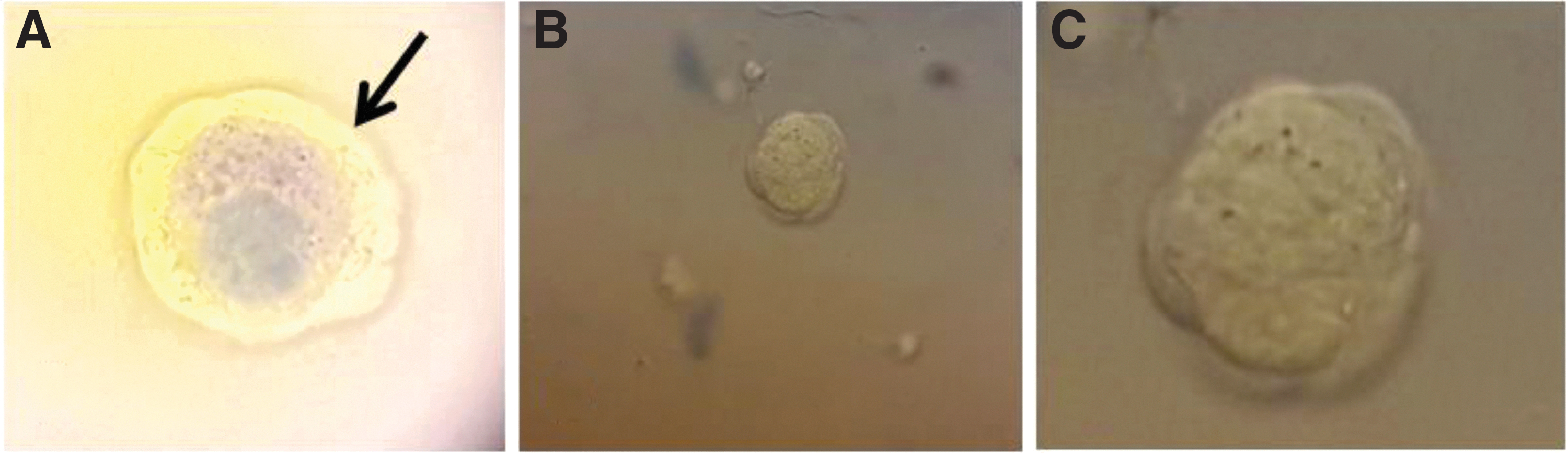
Follicles stained with Trypan Blue at magnification 10
Evaluation of apoptosis
The primary antibody used was a monoclonal rabbit anti-cleaved caspase-3 antibody (9664S; Cell Signaling Technology, Danvers), which is a specific marker of apoptotic cells, the secondary antibody was FITC-conjugated goat anti-rabbit IgG (Cell Signaling Technology). The positive control used was a section of sheep thymus, which is a tissue with a high rate of apoptosis (Fig. 4). OT was incubated 2 h after slow freezing/thawing and vitrification/warming for the study of apoptosis. OT sections were deparaffinized in methylcyclohexan (Sigma-Aldrich) and absolute ethanol (Sigma-Aldrich), then rinsed in water. The unmasking was done by heating for 15 min in immersion in sodium citrate pH = 6 (Merck, Darmstadt, Germany).
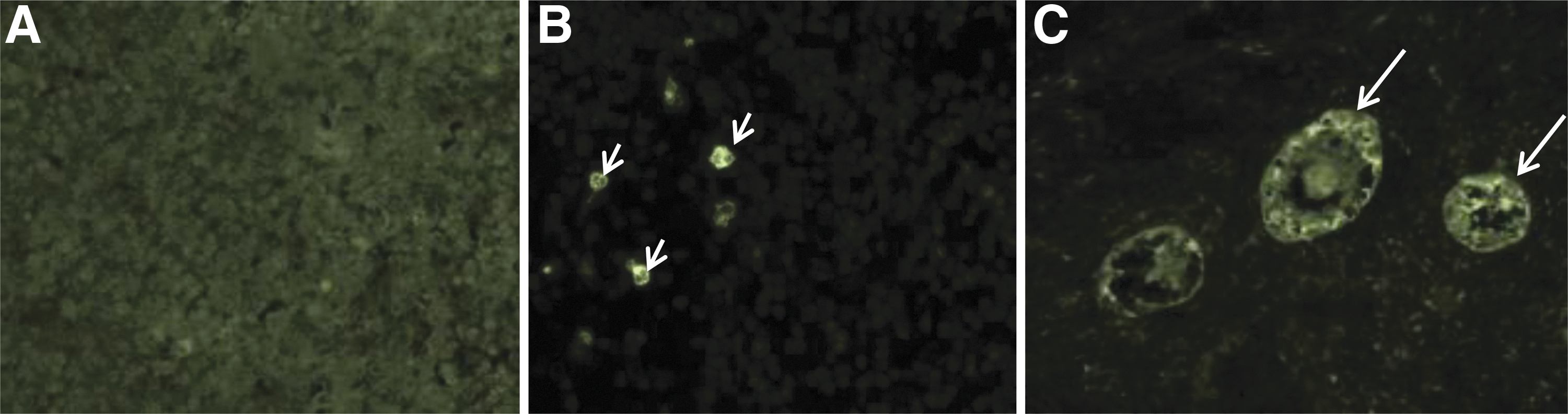
Evaluation of apoptosis on section of thymus at magnification 10
After blocking, slides were then incubated overnight at 4°C in a humid chamber with anti-cleaved caspase-3 antibody (dilution 1:200). After slides were washed in PBS, they were incubated for 1.5 h in a humid chamber with the secondary antibody (dilution 1:500); propidium iodide (Sigma-Aldrich) was used as a nuclear counterstain. Samples were mounted using an aqueous mounting medium (Fluoprep®; bioMérieux) and slides were kept at 4°C, in a dark and humid chamber until reading. Two readers independently observed the slides using fluorescent microscopy (Axiovert 200M; Carl Zeiss, Oberkochen, Germany); fluorescence was dichotomized to no stain (negative expression of cleaved caspase-3) and strong fluorescence (intensity level >50%, positive expression) (Fig. 4).
RNA extraction and complementary DNA synthesis for molecular assessment
Total RNA was extracted from fresh OT, frozen/thawed OT, and vitrified/warmed OT using the RNeasy® Plus Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. We used the whole warmed tissue. The RNA samples were treated with DNase to remove any genomic DNA contamination before proceeding to complementary DNA (cDNA) synthesis. RNA was eluted twice in 30 μL of RNase-free water after extraction and consecutively collected in a final volume of 30 μL and was conserved at −80°C. The RNA concentration was determined by spectrophotometry; the ratio of the readings at 260 and 280 nm (A260/A280) provided an estimate of the purity of RNA.
A total of 500 ng of the extracted RNA was used for cDNA synthesis using the High-Capacity RNA-to-cDNA® Kit (Applied Biosystem, Waltham, MA). The cDNA synthesis was performed at 37°C for 60 min and stopped by heating to 95°C for 5 min. The obtained cDNA was stored at −20°C and ready for real-time polymerase chain reaction (PCR) analyses.
Real-time reverse transcription polymerase chain reaction
The primers for real-time reverse transcription PCR (RT-PCR) were found by literature review (Table 1).30–33 Each primer was verified by using the University California, Santa Cruz (UCSC) Genome browser to check their specificity, target region, and size. Only verified primers were used for the PCR analyses. A total of seven genes were analyzed: anti-Müllerian hormone (AMH), cytochrome p450 family 11 subfamily A member 1 (CYP11A), steroidogenic acute regulatory protein (STAR), growth differentiation factor 9 (GDF9), zona pellucida glycoprotein 3 (ZP3), cyclin D2 (CCND2), and cyclin-dependent kinase inhibitor 1A (CDKN1A). We chose to evaluate GDF9 and ZP3 because they play a dominant role in follicular development, they code for proteins that are factors produced by the oocyte. AMH, CYP11A, and STAR encode proteins secreted by granulosa cells; CYP11A and STAR are essential for the proper functioning of steroidogenesis and AMH plays a major role in folliculogenesis by controlling the exit of follicular stock and has an indirect effect on the growth of preantral follicles by inhibiting the action of FSH. Finally, CCND2 and CDKN1A are involved in cell cycle regulation. CCND2 is involved in ovarian granulosa and germinal cell proliferation. CDKN1A encodes an inhibitory protein to regulate cell growth and cell response to DNA damage.
Oligonucleotide Primer Sequences for Polymerase Chain Reaction
AMH, anti-Müllerian hormone; CCND2, cyclin D2; CDKN1A, cyclin-dependent kinase inhibitor 1A; CYP11A, cytochrome p450 family 11 subfamily A member 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GDF9, growth differentiation factor 9; STAR, steroidogenic acute regulatory protein; ZP3, zona pellucida glycoprotein 3.
One-step RT-PCR was performed using the StepOneplus real-time thermal cycler (Applied Biosystems) and using the Quantitect SYBR Green RT-PCR Kit (Thermo Fisher, Waltham). The reference gene was glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Before quantitative analysis, optimization procedures were performed by running real-time RT-PCR with or without a template to verify the reaction conditions, including the annealing temperatures of the primers and specific products. The real-time thermal conditions included a holding step at 50°C for 2 min and 95°C for 2 min, a cycling step at 95°C for 15 s, 60°C for 15 s, 72°C for 15 s, followed by a melt curve step 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. Each sample was analyzed in triplicate; water was used as negative control.
Statistical analysis
The analysis was performed with the R software (R Core Team 2014). To compare nonindependent proportions a Generalized Linear Mixed Model (GLMM) was used. By this way, the ratios estimated by the GLMM were not a simple division of two values but the inverse of a logit function. However, the estimated ratios by the GLMM were near the ratios calculated by a simple division. Moreover, to take account of the overdispersion of the values, a quasibinomial distribution was used for the calculation and the test of the difference among the ratios. Thus, a precise analysis was performed and a correct way of calculation of ratios was used. To perform this study the “glmmPQL” function of the MASS package was used. The Kruskal/Wallis nonparametric test, was used to analysis the results of real-time RT-PCR data. A test was considered statistically significant when p-value was under 0.05.
Results
Proportion of intact follicles
For primordial follicles, the slow freezing protocol had a significant negative impact on the proportion of intact follicles compared with fresh tissue (37.6% vs. 62.5%, p = 0.003), whereas the vitrification protocol did not have a significant impact (56.0% vs. 62.5%, p = 0.375; Table 2). However, there is a trend toward a decrease in this proportion after vitrification compared with fresh tissue. The proportion of intact primordial follicles was higher for vitrification compared with slow freezing and this effect was statistically significant (p = 0.037).
Impact of Cryopreservation Protocol Compared with Fresh Ovarian Tissue on Morphology of Follicles According to Developmental Stage
p-Value for slow freezing versus fresh, value for correspond.
p-Value for vitrification versus fresh.
CI, confidence interval; OT, ovarian tissue.
For primary follicles, slow freezing had a significant negative impact on the proportion of intact follicles compared with fresh tissue (25.7% vs. 89.7%, p = 0.001), as did vitrification (60.8% vs. 89.7%, p = 0.004; Table 2). In addition to that, when comparing the proportion of intact primary follicles between the two cryopreservation techniques, more intact follicles after vitrification are observed compared with slow freezing (p = 0.003).
For secondary follicles, slow freezing had a significant negative impact on the proportion of intact follicles compared with fresh tissue (78.8% vs. 96.1%, p = 0.035), whereas the vitrification protocol did not have a significant impact although there was a trend toward a decrease of this pool as well (83.6% vs. 96.1%, p = 0.073; Table 2).
Vitality of follicles
As compared with fresh tissue, there was no significant impact of slow freezing (84.9% vs. 80.4%, p = 0.145) or vitrification on follicular vitality (82.9% vs. 80.4%, p = 0.403; Table 3). The proportion of living follicles was similar between the two cryopreservation techniques (p = 0.350).
Impact of Cryopreservation Protocol Compared with Fresh Ovarian Tissue on Follicles’ Vitality
p-Value for slow freezing versus fresh, value for correspond.
p-Value for vitrification versus fresh.
Apoptosis of follicles
The proportion of apoptotic primordial follicles was significantly greater than in fresh tissue after both slow freezing (12.6% vs. 5.1%, p = 0.009) and vitrification (13.9% vs. 5.1%, p = 0.003); as was the case for primary follicles (54.4% vs. 9.1%, p = 0.010, and 36.1% vs. 9.1%, p = 0.004, respectively). The effect on secondary follicles was more difficult to interpret owing to the small number in each group; there was a trend toward increased apoptosis after slow freezing (Table 4).
Impact of Cryopreservation Protocol Compared with Fresh Ovarian Tissue on Follicles’ Apoptosis According to Developmental Stage
p-Value for slow freezing versus fresh, value for correspond.
p-Value for vitrification versus fresh.
Gene expression
Concerning the genes expressed by the granulosa cells, the mean fold change ± SEM of AMH messenger RNA (mRNA) was 2.00 ± 1.07 in frozen OT and this was 0.87 ± 0.80 in vitrified OT; there was no significant difference between vitrified OT and fresh tissue (p = 0.71) and a trend toward overexpression after slow freezing (p = 0.07). The mean ± SEM fold change of CYP11A mRNA was 0.70 ± 0.71 in frozen OT and 2.96 ± 0.93 in vitrified OT; there was no significant difference between frozen OT and fresh tissue (p = 0.66) and the change after vitrification was significantly greater (p = 0.003). The mean ± SEM fold change of STAR mRNA was 0.50 ± 0.51 in frozen OT and this was 1.92 ± 1.91 in vitrified OT; there was no significant difference between frozen OT and fresh tissue (p = 0.3) and a trend toward overexpression after vitrification (p = 0.06).
Concerning the genes expressed by the oocytes, the mean fold change ± SEM of GDF9 was 1.63 ± 1.91 in frozen OT and this was 1.68 ± 1.92 in vitrified OT; there was no significant difference between cryopreserved OT and fresh tissue (p = 0.46 and p = 0.25). The mean ± SEM fold change of ZP3 mRNA was 3.12 ± 2.80 in frozen OT and this was 2.00 ± 2.07 in vitrified OT; there was no significant difference between cryopreserved OT and fresh tissue (p = 0.12 and p = 0.29).
Concerning cell cycle genes, the mean fold change ± SEM of CCND2 was 0.39 ± 0.56 in frozen OT and this was 1.39 ± 0.79 in vitrified OT; the change after slow freezing was significantly lower (p = 0.04), while there was no significant difference between vitrified OT and fresh tissue (p = 0.28). The mean ± SEM fold change of CDKN1A mRNA was 0.70 ± 0.71 in frozen OT and this was 2.50 ± 3.05 in vitrified OT; there was no significant difference between cryopreserved OT and fresh tissue (p = 0.1 and p = 0.14; Fig. 5).
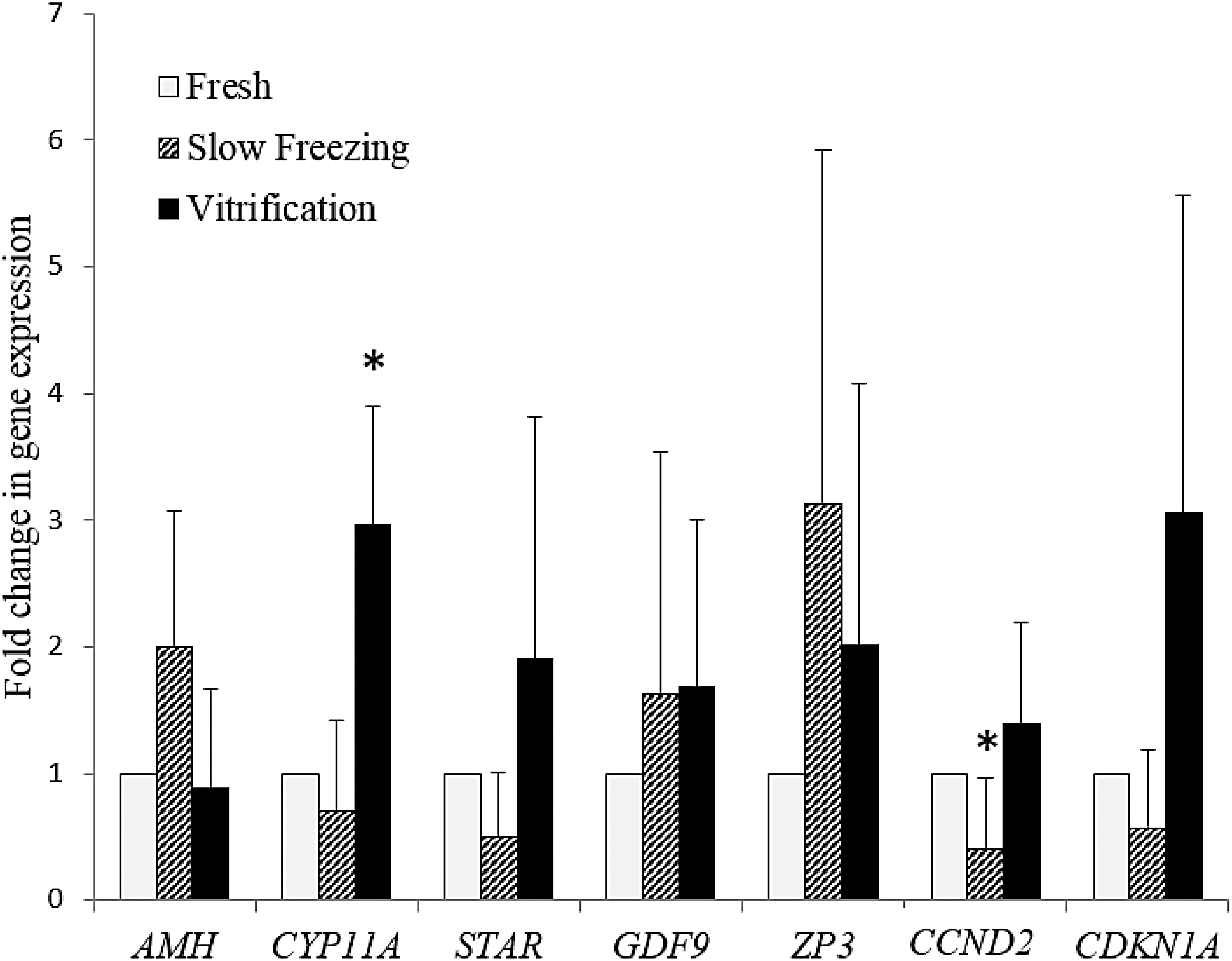
Change in gene expression as measured by messenger RNA extraction from OT and polymerase chain reaction analysis. The value of the expression of each gene in fresh tissue was set at 1; results are presented as mean ± SEM; *p < 0.05 with respect to fresh OT. AMH, anti-Müllerian hormone; CCND2, cyclin D2; CDKN1A, cyclin-dependent kinase inhibitor 1A; CYP11A, cytochrome p450 family 11 subfamily A member 1; GDF9, growth differentiation factor 9; STAR, steroidogenic acute regulatory protein; ZP3, zona pellucida glycoprotein 3.
Discussion
Cryopreservation of OT is now a recognized method of preserving female fertility but the choice of technique, slow freezing or vitrification, remains a dilemma for biologists. In the present study, slow freezing had a significantly negative impact on the primordial follicle morphology compared with fresh tissue, which was not found for vitrification. We also observed a significantly lower proportion of intact primary follicles after vitrification and slow freezing compared with fresh tissue. It is also interesting to note that, when compared with slow freezing, vitrification had a significantly lower impact on follicle morphology. These results are consistent with only two studies,6,34 while only four studies indicated a significantly higher proportion of intact follicles10–12,14; the others observed no significant difference between cryopreservation techniques.3–5,7,8,15,17,18,20–22,35 However, the heterogeneity of the vitrification protocols, of cryoprotectants used, of the size of ovarian fragments, and the methodology used in studies investigating morphology after cryopreservation precludes any meaningful interpretation. 36 For instance, the criteria for evaluating morphology were different and the comparison was not always made to the same reference (either between slow freezing or vitrification and fresh tissue, or between slow freezing and vitrification). It is also of note that the overwhelming majority of such studies, including the present one, investigated follicular morphology immediately after warming.3,5–7,9,14–17 However, a minority included a culture step after warming, but the results were also contradictory; some reported no significant difference between slow freezing and vitrification,21,22,37 while others a significant difference one way or the other.12,13,35,38
As compared with fresh tissue, there was no significant impact of slow freezing or vitrification on follicular vitality. The large proportion of living follicles is reassuring and supports the potential repair capacity of the ovary. Despite morphological alterations of the follicles, they remain alive and therefore potentially capable of developing during grafting. It is important to note that morphometric analysis immediately after cryopreservation may not truly reflect follicle development potential. 39 Herein, follicle apoptosis was significantly more frequent after vitrification and slow freezing compared with fresh tissue. This is in accordance with the majority of previous reports that found a significant increase in apoptosis in granulosa cells that were frozen compared with fresh OT.7,40 In addition, we did not find any significant difference in follicular apoptosis between vitrification and slow freezing. In the literature, only two studies conducted by the same team showed that vitrification caused fewer alterations in DNA fragmentation than slow freezing.6,8 Other studies showed that there was no difference between the two cryopreservation techniques.3,5,10,16,35
It would be interesting to evaluate the proportion of apoptotic follicles after tissue culture to evaluate the repair capacities. Apoptosis would be a much better marker of competence of primordial follicles after cryopreservation than follicle morphology, 40 which, according to the results obtained in the present study, would lead to the conclusion that vitrification would be equivalent to slow freezing.
Taken together, these results indicate that the vitrification protocol used herein was at least as effective as slow freezing but do not allow to conclude which technique was better. The investigation of gene expression in different cell types that compose OT could provide additional data and it is of note that in the present study vitrification of ovine OT had less of a negative impact than slow freezing. Few studies have investigated the impact of cryopreservation at the molecular scale on animal models.
We studied the impact of OT preservation techniques on oocytes by evaluating the expression of GDF9 and ZP3 genes. GDF9 is an oocyte factor that plays an important role during folliculogenesis. 41 We did not observe any significant difference between fresh and frozen or vitrified tissue. This result is also observed on human OT 42 and mice, 43 and therefore it can be concluded that oocyte development competence is not negatively affected by cryopreservation. ZP3 is also important for zona pellucida during folliculogenesis (Hasegawa, 2009). We did not observe a significant difference between fresh and frozen or vitrified tissue, although there was a trend toward ZP3 overexpressed in cryopreserved tissues. Wang et al. did not observe the same effect; ZP3 was underexpressed after slow freezing and vitrification and the authors concluded that the zona pellucida was altered after cryopreservation, explaining higher apoptosis rates in these tissues compared with fresh tissue. 42
To evaluate the paracrine hormonal environment of OT, we studied the expression of the AMH gene as well as the steroidogenesis genes CYP11A and STAR. AMH is secreted by granulosa cells from growing follicles. We did not observe any significant difference but it is interesting to note that there was a trend toward overexpression of this gene after slow freezing. Wang et al. reported no significant difference between frozen and fresh tissue but they observed an underexpression after vitrification. It is difficult to interpret the results reported by Wang et al. because their study of gene expression was carried out after in vitro follicle culture, 42 which is known to decrease gene expression. 31
Herein we studied two genes of the cell cycle CDKN1A and CCND2. An increase in CDKN1A leads to an increase in cellular apoptosis. On the other hand, a decrease in its expression leads to an increase in cell proliferation. 44 We observed a trend toward underexpression after slow freezing and overexpression after vitrification, however, we also found that apoptosis of follicles was significantly more frequent after both slow freezing and vitrification than in fresh tissue. This seems to suggest that the apoptosis observed after slow freezing was related to another mechanism, and could also be partly responsible for the apoptosis of follicles after vitrification, but also that underexpression of CDKN1A after slow freezing did not protect against apoptosis. Slow freezing also significantly decreased the expression of CCND2, which is a cyclin with an important role in the proliferation of germ cells and granulosa cells. Such a decrease in CCND2 expression, and therefore in cell proliferation, may play a part in the slow recovery of the graft after transplantation, which is reported to be between 4 and 6 months after transplantation.45–47 This is commonly explained in the literature to be related to the time needed for revascularization of the tissue, 48 but the present study suggests that this may also be related to the alteration of granulosa cells. It would therefore be of interest to explore gene expression and cell repair capacity in frozen and vitrified OT grafts.
Conclusion
Vitrification preserved follicular morphology better than slow freezing and led to gene overexpressed, while slow freezing led to gene underexpression. The results provide additional arguments for the value of using vitrification, but do not make it possible to clearly decide in favor of one of the two techniques. To this end, two studies are being conducted in parallel: the grafting of vitrified and frozen OT using the ovine model to evaluate the functionality of cryopreserved tissue, and the application of this vitrification protocol to human OT with evaluation at three different times, after thawing as in this study and then after 2 and 6 days of in vitro culture.
Footnotes
Acknowledgments
The authors thank Philip Robinson (DRCI, Hospices Civils de Lyon) for help in article preparation. They also thank their many collaborators who have been involved in their study.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received.