
Abstract
In heart failure, cardiac fibrosis is the result of an adverse remodeling process. Collagen is continuously synthesized in the myocardium in an ongoing attempt of the heart to repair itself. The resulting collagen depositions act counterproductively, causing diastolic dysfunction and disturbing electrical conduction. Efforts to treat cardiac fibrosis specifically have not been successful and the molecular etiology is only partially understood. The differentiation of quiescent cardiac fibroblasts to extracellular matrix-depositing myofibroblasts is a hallmark of cardiac fibrosis and a key aspect of the adverse remodeling process. This conversion is induced by a complex interplay of biochemical signals and mechanical stimuli. Tissue-engineered 3D models to study cardiac fibroblast behavior in vitro indicate that cyclic strain can activate a myofibroblast phenotype. This raises the question how fibroblast quiescence is maintained in the healthy myocardium, despite continuous stimulation of ultimately profibrotic mechanotransductive pathways. In this review, we will discuss the convergence of biochemical and mechanical differentiation signals of myofibroblasts, and hypothesize how these affect this paradoxical quiescence.
Impact statement
Mechanotransduction pathways of cardiac fibroblasts seem to ultimately be profibrotic in nature, but in healthy human myocardium, cardiac fibroblasts remain quiescent, despite continuous mechanical stimulation. We propose three hypotheses that could explain this paradoxical state of affairs. Furthermore, we provide suggestions for future research, which should lead to a better understanding of fibroblast quiescence and activation, and ultimately to new strategies for the prevention and treatment of cardiac fibrosis and heart failure.
Introduction
The most prevalent reason for cardiovascular death is heart failure, which has a 5-year mortality rate of 50%. 1 Heart failure is initiated by damage to cardiomyocytes or the extracellular matrix (ECM), which sets off an everlasting structural remodeling process in the myocardium. This adverse remodeling causes an altered geometry and function of the left ventricle as a result of cardiomyocyte hypertrophy, cardiomyocyte death, myofibroblast activation, and myocardial fibrosis.2,3 The deposition of collagens in the ECM by myofibroblasts defines cardiac fibrosis.4–6
Excessive ECM deposition is initially a repair mechanism to maintain structural integrity, but eventually leads to a reduced function of the heart. The increased collagen content of the myocardium results in a fibrotic and stiffening myocardium, and thereby leads to diastolic dysfunction.7,8 Furthermore, fibrotic tissue impairs the electrical conduction in the heart, increasing the risk of fatal arrhythmias. 9
Despite a surge in cardiac fibrosis-related research, no surgical or pharmacological treatment exists that targets cardiac fibrosis specifically. Some drugs intervening in systemic blood pressure have slowed down or reversed cardiac fibrosis. 9 Whether this is a direct effect of the drug on cardiac fibroblasts or an indirect mechanobiological effect mediated through less myocardial wall stress due to lower blood pressure remains unknown. To target cardiac fibrosis specifically, the cellular and molecular etiology of profibrotic processes must be fully understood.
An abundance of biochemical molecules has been identified to play a role in cardiac fibrosis. Transforming growth factor beta (TGF-β) is a well-established regulator of the fibrotic process, but angiotensin II (AngII), endothelin-1, and platelet-derived growth factor (PDGF), as well as several microRNAs have been found to be involved.10,11 Besides the biochemical cues, the mechanical forces acting on cardiac tissue also play a role in the development of fibrosis. Cardiac fibroblasts have mechanosensitive pathways that can induce profibrotic gene expression. 12 Considering that the heart is a highly dynamic organ, the question arises how cardiac fibroblasts in the healthy myocardium remain quiescent, despite continuous stimulation of mechanotransductive profibrotic pathways. This review aims to address this paradox.
Cardiac Fibroblasts
Cardiac fibroblasts
Cardiac fibroblasts comprise around 15% of the non-cardiomyocyte population in the adult myocardium. 13 They regulate the homeostasis of the ECM by producing matrix components such as collagen type I and III, proteoglycans, glycosaminoglycans, and glycoproteins. 14 In a physiological situation, cardiac fibroblasts in the myocardium retain a quiescent phenotype. However, upon cardiac injury, cardiac fibroblasts undergo a phenotypical conversion to myofibroblasts in response to proinflammatory cytokines, growth factors, and mechanical signals. 15
Myofibroblast activation
Cardiomyocyte death, pressure overload, and myocardial inflammation are the most common triggers of tissue injury in the myocardium. Subsequently, the inflammatory response induces repair processes consisting of an inflammatory, proliferative, and maturation phase. 9 In the early inflammatory phase, immune cells are attracted to the site of injury to clear away cell debris and form a provisional fibrin matrix. During the proliferative phase, the proinflammatory signaling is dampened by various subsets of immune cells and activated fibroblasts start depositing a collagen-based matrix. 16 In the maturation phase, the deposited matrix matures into a dense collagen network and fibroblast activation diminishes.16–18
The activation of cardiac fibroblasts into proliferative, contractile, ECM-depositing myofibroblasts is the hallmark of cardiac fibrosis. 9 Activated fibroblasts are found in abundance in diseased cardiac tissue and display an extensive endoplasmic reticulum, which enables them to secrete large amounts of ECM components, such as collagen type I, nonfibrillar collagen type IV, thrombospondin-1, ED-A variant fibronectin, and periostin.15,19,20
The contractile features that enable, among other functions, fibroblast migration to the site of injury are a result of actin-containing stress fiber formation in the cytoplasm. The incorporation of α-smooth muscle actin (α-SMA) has long been regarded as the defining characteristic of the myofibroblast phenotype. However, not all activated fibroblasts express α-SMA in their stress fibers and α-SMA expression eventually decreases in the late phases after injury.9,12,16–18 Recent studies have shown that quiescent fibroblasts are activated upon injury and a majority of those activated fibroblasts will express α-SMA temporarily. From 7 to 10 days after injury, the activated fibroblasts will stop expressing α-SMA and transition to a new fibroblast state, which is suited for the environment of a maturing scar. This fibroblast state has been described as the matrifibrocyte or the homeostatic-like fibroblast and has a unique gene signature, which shows some similarities with chondrocytes, osteoblasts, and quiescent fibroblasts. 18
The origin of cardiac (myo)fibroblasts
Resident cardiac fibroblasts in the adult myocardium are derived from endothelial-to-mesenchymal transition (EndMT) and epithelial-to-mesenchymal transition (EMT) in the fetal heart. 21 These processes consist of cells migrating into the myocardium, transforming into mesenchymal cells, and losing their endothelial and epithelial signatures. Closely related to resident cardiac fibroblasts are the fibroblast-like cells of the heart valves—valvular interstitial cells (VICs)—which migrate toward the valvular areas in the developing heart. 21
Upon injury in the adult heart, resident cardiac fibroblasts are activated, but several other cell populations could also act as precursors to myofibroblasts.12,22 Pericytes, vascular smooth muscle cells, and circulating fibrocytes and other myeloid progenitors can all differentiate into myofibroblasts.23–32 Furthermore, injury to the heart could lead to reactivation of EndMT and EMT, adding endothelial cells and epicardial cells to the pool of proposed activated fibroblast precursors.21,22,33,34 The relative contribution of each of these precursors to the activated fibroblast population in the injured heart has been subject of fierce debate. However, recent lineage-tracing experiments convincingly show that activated fibroblasts primarily differentiate from resident cardiac fibroblasts in cases of pressure overload and myocardial infarction and that the contribution of vasculature-associated cell types, epicardial cells, and circulating cells is minimal.17,18,35–37 And although not all resident cardiac fibroblasts respond to injury identically, these lineage-tracing experiments revealed the response to injury of fibroblasts of epicardial and endothelial origin to be the same.35,36
Cardiac fibroblast and myofibroblast identification
Historically, most studies of cardiac fibroblasts have taken place in vitro, as these cells were classically isolated from cardiac tissue through their capacity to adhere to tissue culture plastic. 16 Characterization of these cell populations relied on markers that were only partially specific for cardiac fibroblasts. Vimentin, fibroblast-specific protein-1 (FSP1), and discoidin domain-containing receptor 2 (DDR2) have all been used as markers for fibroblasts, despite also being expressed by other cardiac cell types. Similarly, α-SMA has traditionally been used as a marker for the myofibroblast state, even though vascular smooth muscle cells and pericytes also express α-SMA.16,21,38
The aforementioned recent lineage-tracing and single-cell sequencing studies have provided more clarity on the different cell populations of the heart.17,18,35–37 This has also led to more consensus on the definition and markers of cardiac fibroblasts. Tcf21 and PDGFRα expression define the cardiac fibroblast population in the absence of endothelial, hematopoietic, and vascular smooth muscle cell markers. Periostin and ED-A fibronectin are expressed by activated fibroblasts, whereas α-SMA is expressed by the myofibroblast fraction of activated fibroblasts. 38
Molecular Physiology of Cardiac Fibrosis
A complex interplay of local chemokines, growth factors, cytokines, hormones, and systemic factors compile the biochemical factors involved in fibrogenic signaling in cardiac tissue. In heart failure patients, many blood-borne markers are associated with cardiac fibrosis, although it remains difficult to link these circulating markers to causal upregulations of local myocardial protein expression. Moreover, most of the systemic factors include blood pressure-altering hormones, which can indirectly influence fibrosis through mechanical pathways, rather than activating cardiac fibroblasts directly. 14 The biochemical signaling cascades are highly converged, with the TGF-β superfamily and its pathways as central regulators of the fibrotic process (Fig. 1).
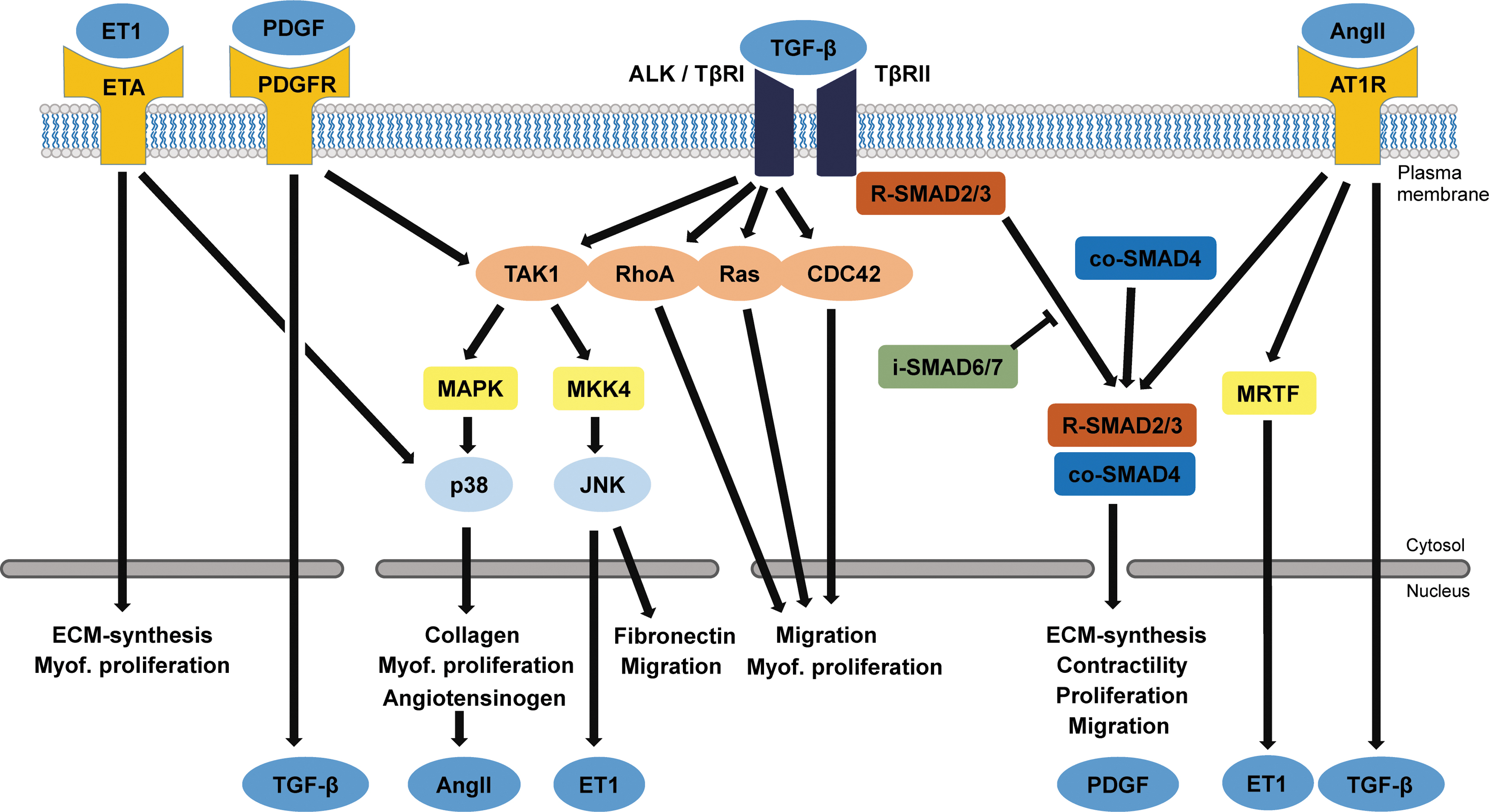
Biochemical signals and intracellular pathways leading to fibrosis. The main actuator of fibrosis is TGF-β, which binds to TβRII. Upon ligand binding, this receptor forms a complex with TβRI (also known as ALK) to activate SMAD signaling through phosphorylation of R-SMAD2/3, which then translocates with co-SMAD4 to the nucleus to activate transcription of profibrotic genes. Aside from the canonical SMAD-dependent pathway, TGF-β can exert profibrotic effects through the TβRI. This noncanonical pathway has many effectors, including TAK1 and small GTPases. TAK1 activates the p38/MAPK pathway, which not only induces collagen and angiotensinogen production but is also an activator of myofibroblast proliferation. This pathway is potentiated by association of ET1 to the ETA, which also directly activates ECM synthesis and myofibroblast proliferation. The PDGF pathway not only positively influences the noncanonical TGF-β pathway by activating TAK1 but can also activate secretion of TGF-β. Binding of AngII to the AT1R positively influences SMAD2/3 signaling, promotes ET1 expression through MRTF, and can drive expression of TGF-β. ECM, extracellular matrix; TGF-β, transforming growth factor beta. Color images are available online.
Canonical (SMAD dependent) TGF-β signaling
TGF-β superfamily members activate the TGF-β type II receptor (TβRII), which in turn transphosphorylates the intracellular domain of TGF-β type I receptor (TβRI), which is also known as activin receptor-like kinase (ALK). 39 Currently, seven subtypes of these ALKs are recognized, of which two are relevant in cardiac fibroblasts: ALK5 and ALK1.40,41 Intracellular receptor-associated Smads (R-Smad2 and R-Smad3) are direct targets of ALK5, and once activated form a trimeric complex with Smad4. This complex translocates to the nucleus where it activates a profibrotic gene program, yielding expression of TIMPs, various types of collagen, and α-SMA. 42
On the other hand, ALK1 activation results in activation of Smad1, 5, and 8, leading to attenuation of fibrosis. Heterozygous ALK1 mice showed increased cardiac fibrosis after transverse aortic constriction compared to wild-type mice, and ALK1 has been shown to be downregulated in human heart failure patients. 43 An interesting modulator of ALK1 and ALK5 signaling is endoglin, which promotes profibrotic signaling through ALK5 and decreases antifibrotic signaling through ALK1. 44
Furthermore, inhibitory Smads antagonize TGF-β signaling by either blocking the phosphorylation of R-Smad3 or blocking the dimerization with Smad4. 45 Smad6 expression is induced by Smad1 and 5, whereas Smad7 expression is induced by Smad3. 5
Activation of these pathways relies on the activation of the TGF-β type I and II receptors, which can be activated by members of the TGF-β superfamily. This cytokine system consists not only of the three isotypes of TGF-β but also of bone morphogenic proteins (BMPs) and growth and differentiation factors (GDFs), which make up more than 30 ligands in total.40,41,44 TGF-β1 is the most widely studied TGF-β superfamily member in cardiac fibrosis and primarily exerts profibrotic actions through the ALK5/Smad2/3 pathway, whereas BMP9 and BMP7 have recently been shown to exert their antifibrotic effects through the ALK1/Smad1 and ALK2/Smad1 pathway, respectively.9,44,46,47
Noncanonical (SMAD independent) TGF-β signaling
The SMAD-independent TGF-β pathway involves the activated TβRI, which—apart from activating the canonical pathway—subsequently activates a wide range of other intracellular messengers that make up the noncanonical signaling pathways. The most prominent is TGF-β-activated kinase 1 (TAK1), which exerts profibrotic functions through activation of the p38/MAPK pathway. 48 This induces a rapid transcription of pro-α1(I) collagen 49 and provides proliferative and antiapoptotic signals important in myofibroblast survival. 50 Furthermore, TAK1 activates the MKK4/JNK cascade, driving fibrosis by inducing expression of fibronectin. 51 Some studies indicate that the TβRII directly activates Rho-like GTPases (RhoA, Rac, and CDC4252), which were recently shown to be involved in myofibroblast migration and proliferation.53,54
Non-TGF-β signaling
Angiotensin II
A well-established profibrotic signaling peptide is AngII, which is part of the renin-angiotensin-aldosterone system.9,14 In cardiac fibroblasts, AngII leads to TGF-β expression, proliferation, increased collagen secretion, and an increase in α-SMA stress fiber expression through the AngII type 1 receptor (AT1R).10,55 Moreover, myocardin-related transcription factor (MRTF) activation by AngII leads to endothelin-1 expression, providing a fibrogenic feed-forward mechanism for cardiac fibroblasts. 10 In recent studies, it has been shown that the Hippo pathway member yes associated protein (YAP) can also be activated through AT1R and promotes profibrotic gene expression in cardiac fibroblasts.56–58 Inhibiting AngII signaling is one of the cornerstones of contemporary heart failure treatment.59,60 ACE inhibitors and AT1R blockers have proven to be clinically effective in lowering systemic blood pressures and reducing myocardial fibrosis. 61
Platelet-derived growth factor
PDGF is a family of growth factors, consisting of the isoforms PDGF-A, PDGF-B, PDGF-C, and PDGF-D. In the heart, the main producers of PDGFs are the cardiomyocytes and endothelial cells. PDGFs exert their function through PDGF receptor α or PDGF receptor β (PDGFRα or PDGFRβ). 62
The effects of PDGF in the heart are not fully understood, but the different isotypes seem to have different effects by targeting different cell populations. A study of transgenic mice demonstrates that the cardiac overexpression of PDGF-A results in severely interstitially fibrotic hearts, most likely mediated through resident cardiac fibroblast activation through PDGFRα. The same study shows that cardiac overexpression of PDGF-B results in a markedly milder phenotype in which fibrosis is mainly localized around the vasculature, presumably through activation of pericytes or vascular smooth muscle cells. 63
PDGF induces cardiac fibroblast proliferation and differentiation by activating noncanonical TGF-β pathways such as Ras/MAPK, MKK4/JNK, and p38/MAPK, 64 resulting in the expression of collagen type I, various MMPs and TIMPs, and an increased expression of TGF-β. 65 Furthermore, to complicate this highly integrated crosstalk, TGF-β in turn influences the synthesis of several PDGF isoforms and the expression of PDGF receptors in fibroblasts.10,64–67
Endothelin-1
Endothelin-1 (ET-1) is a soluble, vasoconstrictive peptide released by several cell types, including endothelial cells and cardiac fibroblasts. ET-1 plays a role in aging-related cardiac fibrosis as it prevents apoptosis and stimulates collagen production by altering downstream TGF-β signaling through p38/MAPK.10,68–70 Stimulation of the endothelin-A (ETA) receptor leads to less proliferation, but increased collagen synthesis in human cardiac fibroblasts in vitro. 71
Interleukin-11
Recent literature describes a role for interleukin-11 (IL11) in cardiac fibrosis. RNA sequencing of human cardiac fibroblasts identified IL11 as a highly upregulated protein in response to TGF-β1 stimulation. IL11 expression was found to be highly specific to fibroblasts and it induced fibrotic gene expression in an autocrine manner through ERK-dependent signaling. IL11 knockout mice that were subjected to AngII injections or transverse aortic constriction, both demonstrated less cardiac fibrosis than wild-type mice. IL11 acts downstream of several fibrotic stimuli, including TGF-β, and exerts its effects only on a post-transcriptional level. 72
MicroRNAs
MicroRNAs (miRNA/miR) are small noncoding RNAs of about 22 nucleotides that modulate gene expression by inhibiting translation of target mRNA. Until now, up to 2000 miRNAs have been identified in the human genome, each potentially affecting numerous targets. 73 Several miRNAs have been shown to be involved in regulating myocardial fibrosis, primarily through modification of the pathways described above. MiR-21 modulates the p38/MAPK pathway, inducing cardiac fibrosis and cardiomyocyte hypertrophy.74,75 On the other hand, miR-378 acts as a paracrine inhibitor of the fibrotic response in cardiac fibroblasts by attenuating the profibrotic p38/MAPK pathway, after it has been released by cardiomyocytes in pressure-overloaded hearts. 76 Recently, miR-433 has been shown to be upregulated in both a murine chronic MI model and human heart failure, enhancing fibroblast proliferation and activation. 77 Other known profibrotic miRNAs are miR-22 and miR-34a, whereas miR-1, 15 family, 24, 26a, 29b, 101, 133, and 489 have been found to be antifibrotic. Current knowledge about the role of miRNAs in cardiac fibrosis has been reviewed excellently by Creemers et al. 78
In summary, cardiac fibroblasts are affected by an array of fibrogenic biochemical signals, resulting in an intricate intracellular signaling network that controls proliferation, activation, and eventually matrix deposition. In addition, biomechanical influences can affect these intracellular fibrogenic signaling pathways.
Mechanical Stimuli in Cardiac Fibrosis
Mechanical forces act on fibroblasts through cell-ECM and cell-cell interactions. 79 Cells in the myocardium generally adhere to each other using cadherins and attach to the ECM through integrins. These transmembrane proteins are connected to the cytoskeleton and transduce mechanical signals to the cellular interior, where they are translated into biochemical signaling or have a direct effect on gene expression through modification of chromatin organization and gene accessibility.80,81 Extracellularly, mechanical forces can induce biochemical signaling by activating fibrogenic growth factors in the ECM.
TGF-β release from the ECM upon stretch
TGF-β is stored in the cardiac ECM in a latent form. It is secreted from the cell in a latent complex, in which it is noncovalently bound to latency-associated peptide (LAP). LAP is covalently bound to latent TGF-β binding protein (LTBP), which is part of the cardiac ECM.82,83 Fibroblasts can bind to LAP through αv integrins. Cellular traction on the latent TGF-β complex, generated by the fibroblast's cytoskeleton and conducted through these integrins, induces a conformational change in LAP, which leads to the release of active TGF-β (Fig. 2). 84 The amount of TGF-β released is not only dependent on the force a cell can generate but also on the compliance of the ECM. A higher stiffness of the ECM leads to higher resistance to cellular traction and more TGF-β activation.85,86 This active TGF-β can subsequently activate canonical and noncanonical pathways, leading to fibroblast activation and the expression of contractile α-SMA, thereby providing a positive feedback loop by promoting further cellular contraction and TGF-β release. 12

Force-activated signaling in cardiac fibroblasts leading to profibrotic gene expression. Latent TGF-β is attached to the cells by various integrins in complex with the LAP and with the ECM by RGD-binding LTBP. Upon mechanical strain or degradation of the ECM by MMPs, the latent TGF-β is released and can activate SMAD signaling through the TβII receptor. Integrins also sense forces applied on the cells. Integrin β1 can force dependently activate FAK, which activates profibrotic conversion through MRTF and GTPases such as RHoA, Ras, and CDC42. Activated integrin β1 can positively influence the p38/MAPK pathway upon mechanical strain. This directly induces expression of profibrotic genes, and indirectly mediates profibrotic effects by positively influencing SMAD signaling. Pellino1 (Pel1) is an intracellular force-activated inducer of p38/MAPK signaling, through TRAF6. Integrin β3 can activate the intracellular protein talin upon mechanical strain across the ECM. Talin activates the SMAD-dependent TGF-β pathway through GSK-3β and Src, influencing the fibrotic response. Talin also increases p38/MAPK signaling, which directly affects the conversion of cardiac fibroblasts to myofibroblasts. Finally, when cadherin-cadherin connections are disrupted during cellular migration or mechanical strain, β-catenin translocates to the nucleus to act as a transcription factor for pro fibrotic genes such as fibronectin, laminin, and MMPs. All these mechanically activated pathways have intricate relationships with biochemical signals shown in Figure 1. LAP, latency-associated peptide; LTBP, latent TGF-β binding protein. Color images are available online.
Force-activated integrin signaling modulates fibrosis
A significant amount of in vivo and in vitro evidence supports the theory that integrins are central regulators of fibrotic pathways in cardiac disease, stimulating proliferation, differentiation, and remodeling. 87 Integrins are transmembrane heterodimers consisting of one α and one β subunit and are the main proteins linking the cytoskeleton to the surrounding ECM.
The β1 subunit of integrin binds to extracellular collagen I, and is a force-induced regulator of the fibrotic response in cardiac tissue. Integrin β1 was particularly expressed on cardiac fibroblasts in fibrotic sites. 88 In a mouse pressure overload model, focal adhesion kinase (FAK) was identified as a crucial downstream regulator of integrin β1 signaling in cardiac fibroblasts, mediating proliferation, differentiation, and fibrosis. 89 FAK force-dependently co-localizes with integrin and is activated upon mechanical stress, as well as through TGF-β and PDGF. 87 By inducing translocation of MRTF1 to the nucleus, FAK activates expression of α-SMA. 15 Moreover, the β1 integrin can induce expression of the AngII propeptide angiotensinogen in cardiac fibroblasts, through activation of the p38/MAPK pathway. AngII in turn can promote the expression of β1 and β3 integrins in cardiac fibroblasts in vitro. 87 In conclusion, integrin β1 is a potent mechanosensing protein, activating a range of profibrotic pathways as shown in Figure 2.
The integrin β3 subtype binds to the RGD peptide sequence found on fibronectin and collagen. The expression of this subtype is markedly increased 7 days after infarction, and is essential for deposition of collagen and fibronectin in a pressure overload model. 90 The integrin-associated protein talin orchestrates these effects, by altering signaling through p38/MAPK, Src, and GSK-3β, all of which positively regulate SMAD-independent TGF-β signaling. These pathways ultimately lead to expression of α-SMA and β3 integrin, providing positive feedback leading to phenotypic conversion of cardiac fibroblasts upon mechanical signaling. 91 A recently discovered E3 ubiquitin ligase, termed Pellino1, has been shown to become activated upon mechanical stretch and increase the p38/MAPK pathway through ubiquitinylation of TNF-α receptor association factor-6 (TRAF6). When silenced, Pellino1 causes a reduced TGF-β expression in neonatal rat cardiac fibroblasts as a response to 15% mechanical stretch, as well as a significant decrease in fibrosis and cardiac function in a rat pressure overload model. 92 These results suggest a mechanosensitive role for Pellino1 in cardiac fibrosis.
Another recent finding in cardiac fibroblasts is the involvement of the Hippo pathway in fibrosis.56,93 The Hippo pathway is a developmental pathway that plays an essential role in organogenesis and cell fate. The end effector of this pathway, YAP, has been shown to be regulated by mechanical cues, including cell morphology, attachment, and ECM stiffness. Upon mechanical stimulation, YAP translocates to the nucleus where it enhances profibrotic gene expression, including α-SMA and MRTF.58,94
Adhesion junction mechanoregulation through nuclear β-catenin translocation
Aside from integrins connecting the cells to the ECM, cell-cell connections are anchored by homodimeric cadherin receptors. Intracellularly, these connect to the actin/myosin cytoskeleton with adherens junction (AJ)-associated proteins such as α- and β-catenin, and vinculin. 95 The latter proteins can locally alter the actin network and non-muscle myosin type II (NMMII)-mediated contractility. 96 Inhibition of contractility by the NMMII inhibitor blebbistatin blocks differentiation, 97 suggesting that intracellular contractility is essential for stiffness-dependent differentiation of mesenchymal stem cells.
Upon mechanical disruption of the AJ, β-catenin is released into the cytoplasm, 91 where it is constitutively degraded by a complex of GSK3, Axin, Disheveled (DVL), and the ubiquitin ligase βTrCP. However, when extracellular Wnt binds to the frizzled receptor (Fzd), it sequesters DVL to the plasma membrane, excluding βTrCP from the complex and inhibiting degradation. Stabilized β-catenin can then act as a transcription factor in conjunction with profibrotic co-factors like p300 and CREB-binding protein.98,99 These pathways lead to a mesenchymal phenotype characterized by the expression of fibronectin, laminin, a variety of MMPs, and a downregulation of E-cadherin. 100 Wnt is a key regulator of cardiac, lung, dermal and kidney fibrosis, and exogenous Wnt addition or nuclear accumulation of β-catenin could induce fibrosis in dermal fibroblasts. 98 In VICs, β-catenin is necessary for TGF-β-mediated myofibroblast conversion, and TGF-β can induce stiffness-dependent nuclear translocation of β-catenin. 101 Wnt is upregulated in the epicardium during cardiac injury, and can induce profibrotic gene expression in cardiac fibroblasts. 102 An immortalized cardiac fibroblast cell line showed enhanced migration, expression of collagen type I and III, fibronectin, and α-SMA upon transfections with Fzd, administration of Wnt, or overexpression of β-catenin. 103
Mechanically activated ion channels
Another means of mechanotransduction is through mechanically activated ion channels. Recently, Piezo1, a mechanosensitive nonselective cation channel, has been linked to cardiac fibrosis. Changes in cellular membrane tension lead to the opening of this channel and an influx of calcium ions. Through p38/MAPK signaling, this eventually leads to profibrotic IL-6 expression in both murine and human cardiac fibroblasts. 104
Similarly, mechanically activated ion channel transient receptor potential vanilloid 4 (TRPV4) seems to play an important role in cardiac remodeling and cardiac fibroblast activation. Absence of TRPV4 in a mouse model of myocardial infarction resulted in a decrease in cardiac fibrosis, whereas activation of TRPV4 caused α-SMA and collagen expression through the Rho/Rho kinase/MRTF pathway. 105
In conclusion, systemic regulatory changes, local signaling, and alterations in mechanical environment converge to form proliferative, contractile, and ECM-producing cardiac fibroblasts. Several pathways involved in (non-)canonical TGF-β signaling and non-TGF-β signaling are interpreting and translating a broad range of biochemical and mechanical cues simultaneously. The precise interactions are not fully understood and various mechanotransduction mechanisms still have to be investigated more thoroughly in cardiac fibroblasts for their possible role in fibrosis. 106 Understanding the relationships between biochemical and mechanical pathways calls for appropriate research models in which all these components can be independently controlled.
In Vitro Models to Assess Mechanoregulation in Cardiac Fibrosis
To unravel the precise molecular mechanisms of fibrosis, conventional cell culture and animal models might not suffice. Conventional 2D cell culture does not offer the complexity of the in vivo situation. Animal models, however, do not allow for careful control of biochemical and mechanical conditions of cells, and are not fully recapitulating the human situation due to interspecies differences, lack of comorbidities, and other confounding factors.
Three-dimensional culture platforms have emerged as alternative models, for their closer resemblance to the native cellular environment due to their chemical and biomechanical tunability. These more advanced in vitro models also allow for integration of other relevant biomechanical stimuli that are important in human myocardium, such as cyclic mechanical strain and ECM stiffness.
Controlling ECM properties
The stiffness of the ECM in vivo or the culture substrate in vitro can be sensed by cells, considerably affecting the fibrotic response of fibroblasts. 12 Cells pull on their surroundings through the integrins that connect their cytoskeleton to the ECM. Depending on the encountered resistance, this leads to conformational changes of proteins in the focal adhesion complexes associated with the cytoskeleton, which sets mechanotransductive pathways in motion, which can alter cell behavior. 107 For disease modeling in vitro, it is therefore important to control the mechanical properties of the cell culture substrate, in particular the ECM stiffness.
Hydrogels are water-absorbing polymeric gels that have been extensively used in tissue engineering for their ability to encapsulate cells in a biologically favorable environment. An important advantage of hydrogels is their tunable properties, such as stiffness and elasticity, which can be regulated by modulating the concentration of polymers or cross-linking agents. 108 Their high hydrophilicity in combination with a tunable permeability enables the retention of water, which facilitates the transport of oxygen and nutrients. Moreover, hydrogels with a high resemblance to native ECM often have inherent cell-binding motifs. 109 Their individual biological characteristics, along with their advantages and shortcomings, have been extensively reviewed elsewhere and will not be discussed here.19,110
Using hydrogels with varying stiffness has shown that substrate stiffness has a major effect on myofibroblast differentiation and behavior.111,112 Using a photodegradable synthetic poly(ethylene glycol) hydrogel to lower substrate stiffness during culturing, valvular myofibroblast activation was significantly reduced after light induction, as measured by α-SMA expression, stress fibers, and proliferation. 113 This effect was negated by blocking the PI3K/AKT pathway. 114 Stiffened substrate is paralleled by increased expression of β1 integrin, but no increase in its intracellular mediator FAK. However, the JNK/MKK4 pathway was activated when culturing on polyacrylamide with collagen I, but not on polyacrylamide containing soluble ECM components, indicating that the binding of β1 integrin to collagen is essential for substrate-dependent mechanosensing. 115
Apart from substrate stiffness and natural binding sites, dimensionality also affects cell behavior. Transcriptional profiles of VICs were shown to be markedly different in 3D cultures compared to 2D, especially in genes relating to cell structure and motility. Gene expression in 3D was more similar to native tissue. 116 Furthermore, fibroblasts' focal adhesions to 3D matrix are distinctly different in structure and function in comparison to adhesions in 2D. 117 This could have a profound effect on cellular responses to mechanical stress, as focal adhesions are an important part of the mechanosensing system of the cell.
Mechanical strain effects on cardiac fibroblast differentiation in 2D
The most important force experienced by fibroblasts in the myocardium is the strain caused by the contractility of the cardiomyocytes. Most in vitro studies try to mimic this using deformable membranes on which cells are cultured, enabling static or cyclic strain. 12
As discussed earlier, mechanosensitive pathways can lead to the activation of myofibroblasts. However, the experimental results of the effects of mechanical strain on cardiac fibroblasts vary. Neonatal rat ventricular fibroblasts showed increased synthesis of collagen I, fibronectin, and glycosaminoglycans after 24-h cyclic stretch (Table 1). 118 Cells cultured on elastin also showed increased collagen I production after cyclic mechanical load, which was enhanced by inhibiting p38/MAPK. 119 As this mechanosensing pathway is profibrotic, these results are counterintuitive. Another study stimulated cells on fibronectin-enriched plates and with 10% stretch for 72 h. This decreased TGF-β-induced phosphorylation of R-Smad2, causing a reduction of α-SMA expression, 120 contradicting other studies. Drawing conclusions from these studies is difficult, as the time points, strain magnitudes, and strain frequencies used were not identical.
Overview of Experimental Studies on the Effects of Mechanical Strain on Fibroblast Behavior in Two Dimensional and Three Dimensional
Arrow upwards = increased expression. Arrow downwards = decreased expression.
CF, cardiac fibroblast; COL, collagen; FBS, fetal bovine serum; FN, fibronectin; GAG, glycosaminoglycans; gelMA, gelatin methacryloyl; Hz, Herz; VIC, vascular interstitial cell; ECM, extracellular matrix.
Looking at the available literature, cardiac fibroblasts apparently react very differently to comparable mechanical stimuli under varying biochemical circumstances. The mechanical strain-induced myofibroblast phenotype conversion in vitro is affected by serum content added to culture media. In the absence of serum, rat cardiac fibroblasts stretched on a collagen I-coated surface showed an increase in collagen I and III, which was abrogated by inhibition of PKC or Src. The same experiment in 10% fetal calf serum resulted in a decrease of collagen I and III production. 121
Another factor is the type of matrix to which the cells adhered. It is known that various integrins, with different downstream effects, preferentially bind to different types of ECM, causing a direct relationship between culture substrate and cell behavior. 122 Cells seeded on collagen IV, collagen VI, or laminin, but not on collagen I, showed increased expression of collagen I, collagen III, and α-SMA upon 10% cyclic stretch. Interestingly, cells stretched on collagen I even reduced TGF-β-induced expression of α-SMA and collagen III. 123 To a certain extent, these results elucidate the varying results of the effect of mechanical strain on myofibroblast phenotypic conversion, and exemplify the difficulty of drawing conclusions about this process in vitro.
Mechanical strain effects on fibroblast differentiation in 3D
When fibroblast-like VICs are grown in a 3D collagen matrix, the application of cyclic strain caused a quiescent phenotype with reduced α-SMA expression.124,125 A synergistic increase of α-SMA expression was observed when whole porcine aortic valve leaflets were subjected to a combination of cyclic stretch and TGF-β, whereas cyclic strain alone caused a quiescent phenotype. 126
Cardiac fibroblasts cultured in a 3D system reverted to an α-SMA-negative state with reduced collagen I synthesis when a single static stretch of 30% was applied for 1 week. 127 Another study validated this decrease in myofibroblast markers when cardiac cells were subjected to 5% cyclic stretch for 48 h, with a corresponding reduction in R-SMAD2 phosphorylation. 128
However, a recent study found an increase in cardiac fibroblast proliferation when freshly isolated murine cardiac cells were subjected to 10% cyclic uniaxial strain in a fibrin gel in a microfluidic platform. 129 Yet another recent study partly contradicted these results in a comparable experimental setup. Murine cardiac fibroblasts in a gelatin methacryloyl hydrogel in a microfluidic device that were subjected to cyclic biaxial strains varying from 5% to 20% did not show a relationship between strain magnitude and proliferation rate. An increase in α-SMA, TGF-β, collagen type I, and fibronectin ED-A expression was observed, however. 130
In conclusion, studies investigating the effect of mechanical strain on cardiac fibroblasts are scarce, and generally show contradictory results. The exact strain modes used, the strain magnitude, the cell types used, the biological binding sites, and the mechanical characteristics of the ECM substitute, and possibly other unknown factors all appear to affect the behavior of cardiac fibroblasts and could contribute to the differences between the reported studies. Controlling the microenvironment of cardiac fibroblasts seems to be essential for understanding their behavior.
Discussion
The conversion of cardiac fibroblasts to ECM-remodeling myofibroblasts is the driving cellular mechanism of cardiac fibrosis, and is orchestrated by the converging actions of biochemical and mechanical signals. The contribution of mechanical stimulation to myofibroblast differentiation could explain the limitations of drugs that interfere solely with biochemical signaling; modifying the mechanical cues involved in myofibroblast differentiation is extremely challenging in a tissue as mechanically dynamic as the heart. Intracellular proteins like FAK and talin influence fibrogenic signaling upon mechanical stress through TGF-β-related pathways such as p38, JNK, and Rho-GTPases. A FAK inhibitor was recently found to decrease ECM synthesis by cardiac myofibroblasts in vitro, and partially preserve cardiac function in vivo in a cardiac fibrosis mouse model. 131 This research paves the way for novel therapies targeting mechanoresponsive pathways in cardiac disease, and simultaneously implies the need for an integrated understanding of the intracellular signaling cascades following mechanical and biochemical profibrotic cues.
Approaching the mechanical paradox in cardiac fibrosis
Three-dimensional models are currently being used to study the effect of mechanical stretch on fibroblast behavior. However, difficulties in interpretation arise due to the varying results from different studies. When cardiac fibroblasts are stretched in a 3D environment, they can maintain a quiescent state. This may be intuitive, considering that cyclic strain present in the healthy human myocardium should not lead to cardiac fibrosis. However, the question whether mechanical strain leads to a profibrotic phenotype in cardiac fibroblasts has now reached a paradoxical state: how do mechanically stimulated cardiac fibroblasts remain quiescent in healthy myocardium while their force-activated pathways ultimately lead to expression of profibrotic genes?
We propose three hypotheses, visualized in Figure 3. (1) In healthy myocardium, cardiac fibroblasts orient themselves so as to avoid cyclic strain. Strain avoidance is a principle mechanism for human lung and dermal fibroblasts to stay quiescent and has a similar effect in VICs.124,132,133 However, strain avoidance is a phenomenon that has thus far only been observed in in vitro studies. A recent experimental and computational study found it could not explain cellular orientation by strain avoidance alone, but the strain conditions used in this study are simplified and do not fully recapitulate the motion of the human heart. 134
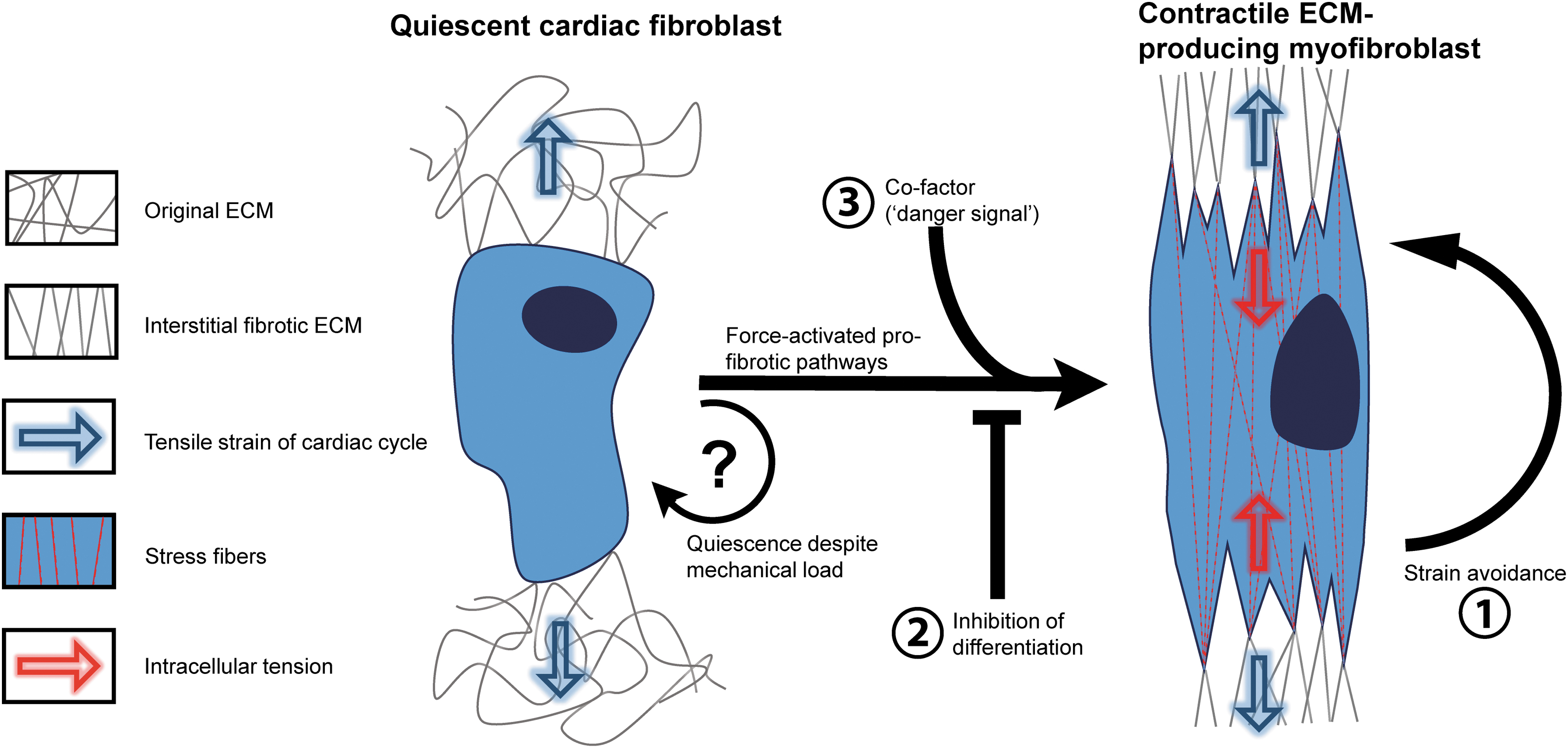
The mechanical paradox in cardiac fibrosis. Cardiac fibroblasts continuously experience a significant mechanical load in the healthy myocardium. During adverse remodeling, the mechanosensitive pathways activate profibrotic signaling, mainly through force-activated integrins. However, these pathways do not induce fibrosis in physiological circumstances. How quiescence in cardiac fibroblasts is maintained is of crucial interest in cardiac fibrosis research. One explanation might be that strain avoidance prevents cardiac fibroblasts from differentiating into myofibroblasts (1). Alternatively, an inhibiting signal might play a role in maintaining quiescence (2) or a profibrotic co-factor might be needed for mechanical induction of fibrosis (3). This could be the TGF-β release from stiff ECM, a reduced supply of nutrients, or other “danger signals” related to cardiomyopathies or injury. Color images are available online.
(2) The mechanical induction of myofibroblast differentiation is actively inhibited in healthy myocardium. Cardiac fibroblasts continuously sense the mechanical strain of their dynamic environment, but the intracellular signaling pathways leading to expression of α-SMA and ECM deposition are inhibited by unknown protective signals. The combination of cyclic stretch and a soft substrate, as present in healthy myocardium, might coincide with intracellular signaling through integrin-mediated FAK and talin to facilitate a quiescent fibroblast state in the nondiseased myocardium. Another possibility is inhibition of the intracellular Smad2/3 complex by Smad6/7 (Figs. 1 and 2). This point of convergence between biochemical signaling and mechanically induced signaling might pose another intracellular inhibition of the myofibroblast differentiation in response to mechanical load in healthy contractile myocardium.
(3) Mechanical stimulation synergistically induces myofibroblast differentiation in diseased myocardium. In other words, cyclic strain only induces fibrosis in cardiac tissue when cells are co-stimulated with a “danger signal.” This hypothesis is supported by the synergistic increase of α-SMA expression in VICs by cyclic uniaxial strain and TGF-β stimulation, while cyclic strain alone causes a quiescent phenotype.124–126 This could implicate that the increased TGF-β signaling during adverse remodeling is crucial for the stress-induced activation of profibrotic genes, which might be explained by the Smad dependence of force-induced integrin β1 and β3 signaling, as shown in Figure 2. The nutrient-depleted and hypoxic environment of cardiac scar tissue and hypertrophic tissue could form other potential co-factors, 20 as reduced serum content also leads to the activation of cardiac fibroblasts in vitro. Other possible co-factors are the different ECM components in the diseased myocardium, to which fibroblasts will bind with different integrins, which have distinct effects on the mechanotransduction pathways in the cell. 122 This could lead to a more prominent role for mechanical cues in diseased myocardium.
It is not necessary for these co-signals to occur at the same time. For instance, prolonged exposure to TGF-β could alter the cellular sensitivity to changes in the mechanical environment. Vice versa, other factors might have a beneficial effect on myofibroblast differentiation and proliferation, depending on the timing of both signals. These findings could have great implications in both fundamental and clinical research in cardiac fibrosis, and can give novel insights in healthy cardiac homeostasis and myofibroblast differentiation in diseased cardiac tissue.
Future perspectives
Our understanding of the cellular pathways interpreting mechanical stress predominantly comes from 2D in vitro culture models. Two-dimensional cyclic stretch devices greatly increased our understanding of the intracellular pathways that link mechanical strain to adaptive signaling. 108 However, in 2D culture models, many factors of the extracellular milieu, such as mechanical cues, cell-cell interactions, and cell morphology, do not adequately mimic the cellular microenvironment in vivo. Therefore, advancement of research to 3D models in cardiac fibrosis is necessary. This will not only be beneficial for fundamental research into novel therapeutics but could also provide alternative means of drug screening and testing. It could partly replace lengthy, costly, and ethically delicate animal testing and could also overcome problems caused by interspecies differences by using human cells.
However, certain challenges remain in creating a physiologically relevant model of cardiac fibrosis. Virtually, all 3D models lack a diversity of ECM, failing to mimic the complexity of the native tissue. 135 An ideal replica of the ECM should not only provide structure and natural binding sites for cells, and have the appropriate mechanical properties but should also contain factors such as periostin, EDA fibronectin, and latent TGF-β complex. Decellularized myocardial ECM has been shown to enhance the differentiation of human embryonic stem cells (ESCs) and cardiac progenitor cells (CPCs) toward cardiomyocytes. 136 Using decellularized myocardial ECM to study cardiac fibrosis in vitro might enhance comparability to in vivo studies, although the limited tunability of its mechanical properties is an important limitation for the use of this material.
Furthermore, the co-culture of other cell types can also impact the fibrotic response in vitro. Recent evidence suggests that cardiomyocytes release soluble factors that affect the proliferative capacity of cardiac fibroblasts.106,137 Conversely, the presence of fibroblasts can also influence the function of cardiomyocytes. Van Spreeuwel et al. demonstrated that increasing the fibroblast number in a 3D co-culture model decreased beating frequency of cardiomyocytes. 138 In addition, our group has shown that co-culture of murine CMs and cardiac fibroblasts in a 3D model of cardiac fibrosis altered mechanical properties and decreased beating rate. 111
Incorporating other cell types into disease-modeling systems will be greatly facilitated by using stem cell-derived cell types. Primary human cardiomyocytes are notoriously hard to maintain in culture and do not proliferate in vitro. 139 Cardiomyocytes derived from ESCs, CPCs, and induced pluripotent stem cells can overcome this hurdle.19,140 Also, endothelial cells and inflammatory cells can be derived from iPS cell lines,141,142 enabling the use of multiple relevant cell types known to play a role in the fibrotic heart.143,144
Finally, controlling and manipulating the biochemical and mechanical microenvironment of in vitro-models is a key aspect of future research. Organ-on-chip technology aims at mimicking functional units of organs in vitro. Making use of microfabrication techniques, microfluidic devices can be developed, which serve as microscale bioreactors. Cells or tissues cultured in these devices can be accurately controlled, both biochemically and mechanically.19,145,146 Combined with the newest developments in cardiac tissue engineering, organ-on-chip technology could offer the right conditions for a step forward in human cardiac fibrosis research.
Conclusion
We introduced a paradox in cardiac fibrosis research, describing how force activation of fibroblasts theoretically should lead to myofibroblast activation, whereas, in fact, quiescence is maintained in healthy myocardium through unknown mechanisms. The development of a model that is mechanically controllable and tunable would greatly advance our understanding of both biochemical and mechanical etiological factors in cardiac fibrosis. More fundamental research is needed to improve our insight in the phenotypic conversion of myofibroblasts that drive fibrosis, and particularly to comprehend the mechanisms that can maintain quiescence, despite mechanical loading.
Footnotes
Disclosure Statement
No competing financial interests exist.
Authors' Contribution
T.B.G. and J.S. wrote and edited the article. D.M., C.B., P.D., A.K., W.S., J.S., and J.H. reviewed the article and provided feedback. All authors agreed on the final version of the article.
Funding Information
We acknowledge the support from Innovation and the Netherlands CardioVascular Research Initiative (CVON): The Dutch Heart Foundation (2017T040), Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science, and the ZonMW Translational Adult Stem Cell grant 1161002016 and Horizon2020 ERC-2016-COG EVICARE (725229).