
Abstract
Bioengineering of skin has been significantly explored, ranging from the use of traditional cell culture systems to the most recent organ-on-a-chip (OoC) technology that permits skin modeling on physiological scales among other benefits. This article presents key considerations for developing physiologically relevant immunocompetent diabetic foot ulcer (DFU) models. Diabetic foot ulceration affects hundreds of millions of individuals globally, especially the elderly, and constitutes a major socioeconomic burden. When DFUs are not treated and managed in a timely manner, 15–50% of patients tend to undergo partial or complete amputation of the affected limb. Consequently, at least 40% of such patients die within 5 years postamputation. Currently, therapeutic strategies are actively sought and developed. However, present-day preclinical platforms (animals and in vitro models) are not robust enough to provide reliable data for clinical trials. Insights from published works on immunocompetent skin-on-a-chip models and bioengineering considerations, presented in this article, can inform researchers on how to develop robust OoC models for testing topical therapies such as growth factor-based therapies for DFUs.
We propose that immunocompetent DFU-on-a-chip models should be bioengineered using diseased cells derived from individuals; in particular, the pathophysiological contribution of macrophages in diabetic wound healing, along with the typical fibroblasts and keratinocytes, needs to be recapitulated. The ideal model should consist of the following components: diseased cells embedded in reproducible scaffolds, which permit endogenous “diseased” extracellular matrix deposition, and the integration of the derived immunocompetent DFU model onto a microfluidic platform. The proposed DFU platforms will eventually facilitate reliable and robust drug testing of wound healing therapeutics, coupled with reduced clinical trial failure rates.
Impact statement
Current animal and cell-based systems are not physiologically relevant enough to retrieve reliable results for clinical translation of diabetic foot ulcer (DFU) therapies. Organ-on-a-chip (OoC) technology offers desirable features that could finally enable the vision of modeling DFU for pathophysiological studies and drug testing at a microscale.
This article brings together the significant recent findings relevant to developing a minimally functional immunocompetent DFU-on-a-chip model, as wound healing cannot occur without a proper functioning immune response. It looks feasible in the future to recapitulate the stagnant inflammation in DFU (thought to impede wound healing) using OoC, diseased cells, and an endogenously produced extracellular matrix.
Background
A wound is simply the loss of skin integrity. 1 When wounds happen in individuals with underlying health conditions (e.g., diabetes, venous insufficiency, etc.), healing is impaired. 2 Wounds that do not heal within 4 weeks are described as chronic.2,3 Chronic wounds include venous leg ulcers (VLU), pressure ulcers, and diabetic foot ulcers (DFU). 4 Chronic wounds are characterized by persistent inflammation and impaired reepithelization 5 (Fig. 1). Therapies and technologies for chronic wound treatment have been developed and tested on animal models.6–8 These latter, indisputably, have provided valuable insights in the area of wound healing, thanks to their physiological complexity. For example, type I and II diabetes can be modeled in a variety of transgenic mouse lines,9–11 and comprehensive investigation of delayed wound healing in vivo at a molecular level can be performed.12–16 However, animal use is progressively becoming less attractive due to irresolvable differences in physiology, metabolism, immunity, and wound healing mechanism between humans and animals –in addition to the distress and discomfort experienced by animals.17–22 Hence, even if a therapy proves effective in animals, it is unlikely to translate seamlessly in humans. In fact, there is a 90% failure rate of therapies shown to be promising in animals when translated into humans.23–25 Furthermore, in a response to the 3R's principle (refinement, reduction, and replacement), which advocates for refinement, reduction, and replacement of animal use for biomedical research, 26 the development of in vitro physiologically relevant wound models has since been an area of growing interest.

Chronic wounds are characterized by persistent inflammation and subsequent impaired wound closure. In chronic wounds, apart from being highly proinflammatory, macrophages are dysfunctional and therefore unable to clear away apoptotic neutrophils, bacteria, pathogens, and cellular debris effectively. Consequently, biofilm formation is favored. Furthermore, the high release of proinflammatory mediators results in increased matrix metalloproteinases to tissue inhibitors of metalloproteinases ratio levels. Hence, excessive ECM breakdown takes place and results in impaired cell–ECM interactions. Finally, KC, although hyperproliferative, is unable to migrate across and reepithelize the wound bed. ECM, extracellular matrix. Reprinted with permission from Wiley. 5
DFU and immune response
DFU are full-thickness chronic wounds below the ankle that can affect the skin, soft tissue, and bone of diabetic individuals. 27 DFU can result from contributing factors of either neuropathy, ischemia, foot deformities, injury, susceptibility to infection, or peripheral arterial/arterial occlusive diseases.28,29 In particular, diabetic peripheral neuropathy results in foot deformity or limited joint mobility. Consequently, uneven pressure distribution and subsequent callus formation at high-pressure zones (at least a 20-fold increase relative to the surrounding skin) occur. When these high-pressure skin zones become exposed to repetitive trauma, local tissue injury occurs; failure to treat the wound effectively in a timely way results in a DFU. 30
In a DFU, neutrophils and monocytes (which differentiate into macrophages in the wound tissue) are recruited slowly, and remain prolonged in the wound bed. 31 The altered cytokine profile released by the first immune cells (neutrophils) makes them dysfunctional and hence renders the wound susceptible to infection. The second-line of immune defence by macrophages has impaired phagocytic activity. Therefore, apoptotic neutrophils, debris, and bacteria are not cleared away effectively from the wound site. 32 Worse still, coupled with insistent inflammation, the hyperglycemic milieu and the presence of advanced glycated end-products (AGE) favor macrophages remaining in their proinflammatory state, rather than transitioning to an anti-inflammatory phenotype favoring wound healing.33–35 Consequently, high levels of proinflammatory cytokines and reactive oxygen species are released. Thereafter, the proliferation phase is marked by an impaired granulation tissue 36 (consisting of a poorly vascularized network surrounded by fibrin cuffs); subsequently, keratinocyte migration and eventually wound closure are hindered. In addition, the production of cytokines and growth factors to recruit fibroblasts and other cells is also hindered. Due to the chronic inflammatory milieu, the remodeling phase is characterized by extracellular matrix (ECM) protein degradation by significantly high levels of matrix metalloproteinases (coupled with reduced expression of tissue inhibitors of metalloproteinases) and aberrant interprotein bonds. Therefore, a mature and organized ECM network cannot be achieved. 31 Overall, nonhealing DFU are characterized by unresolved inflammation and subsequent microbial infection (biofilm formation).
During wound healing, two discrete categories of macrophages are typically involved: M1 and M2 macrophages. M1 macrophages act during the early phase of wound healing by releasing proinflammatory cytokines and “eating up” apoptotic neutrophils, bacteria, dying cells and debris to prevent infection. 33 Thereafter, M2 macrophages facilitate tissue remodeling by releasing matrix metalloproteinases to aid ECM reorganization and growth factors for cell proliferation within the healing tissue.33,37
In DFU, however, macrophages exhibit a dysfunctional phenotype and an imbalance of M1/M2 macrophages. Hence, the ulcer remains stuck in inflammation and cannot progress to the proliferation and re-epithelization phases. 38
Overall, when it concerns the adult organism, wound healing cannot occur without a proper functioning immune response.
In vitro DFU models and limitations
First-generation wound models consisted of monolayers of either FB or KC grown in two dimensions (2D).39,40 A cell-free area (representing a “wound”) is created using either a mechanical, thermal or noncontact approach. 41 Although simple and cost-effective, the lack of cell–cell and cell–matrix interactions, the inability to predict the complex effect of drug metabolism in vivo, and the restricted use of cell types render 2D models quite irrelevant in studying DFU pathogenesis and drug response. For example, KC from diabetic patients is clinically known to have impaired migration 42 ; contrarily, they are likely to migrate when cultured in 2D. 43
Coculture systems have been explored for developing organotypic three dimensional (3D) human skin equivalents (HSE). Three dimensional bioengineered skin constructs for studying DFU healing resulted in more biologically relevant models than predecessors.44–47 HSE generally consist of using hydrogels to embed dermis representative (FB) followed by culturing KC upon the dermal layer at air-liquid interface to initiate epidermal differentiation. Cell-free areas representing wounds are then created, most commonly by using biopunches. 48 Immune cells can be also included to study their paracrine interactions with HSE using Transwell systems. 49 Alternatively, immune cells can be embedded directly into the dermis representative hydrogels with FB.50–53 3D models of DFU reported in the literature include models consisting of FB and endothelial cells (EC) to study angiogenesis in DFU, HSE to study FB and KC interactions, wounded HSE models for reepithelization studies, and self-assembled ECM models.44,47,54
Current 3D models of DFU have demonstrated the vision of engineering the 3D tissue microenvironment in vitro using diseased cells, creating wounds via different techniques, and monitoring cell migration into the cell-free areas. They have provided good understanding to some aspects of DFU pathophysiology, which are clinically observable in DFU patients such as impaired reepithelization, poor angiogenesis, and defective ECM deposition.44,46,47 However, 3D models cannot precisely control spatiotemporal chemical gradients and cannot provide environmental cues (e.g., cyclic stretching), apart from presenting issues related to sampling the lumen contents during pharmacokinetic/pharmacodynamics studies of drugs. 55
Hence, integrating physiological relevant traits is desired for developing reliable in vitro DFU models; the challenges center on optimizing the microenvironment, flow, immune cell mobilization (in response to cytokine/chemokine gradients), and extravasation into the wounded tissue, cyclic stretch, shear stress, fluid to tissue physiological ratios, and noninvasive monitoring by means of robust microsensors.55,56
Immunocompetent skin-on-a-chip models
The inclusion of immune cells remains vital for bioengineering physiologically relevant immunocompetent in vitro models. Blood vessels, apart from supplying oxygen and nutrients to tissues, serve as conduits for immune cell transport to an injured tissue (e.g., DFU) to mount inflammation. 57 Therefore, immune cell mobilization, circulation, and extravasation need to be recapitulated as best as possible using microfluidics. This section will review immunocompetent skin-on-a-chip (SOC) reported in the literature, providing a vision on how DFU may be modeled.
Ramadan and Ting developed a perfusion-based immunocompetent model to study skin barrier function. 58 Distinct monolayers of immortalized human keratinocytes (HaCaT) on the upper chamber and human leukemic monocytes U937-derived dendritic cells on the lower one are divided by a polyethylene terephthalate (PET) membrane (Fig. 2). Following lipopolysaccharide infection, the inflammatory response of the cocultured HaCaT/U937 model was distinct in comparison to both the U937 and HaCaT monocultures; the detected proinflammatory cytokines interleukins (IL)-6 and -1β levels signified that lipopolysaccharide was inhibited from diffusing into the epidermal barrier.
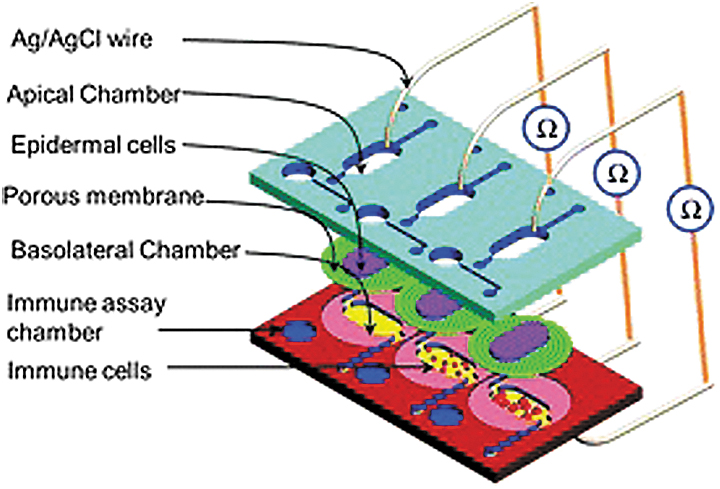
Schematic view of the immunocompetent SOC model incorporating three parallel cell culture chambers and silver/silver chloride (Ag/AgCl) wire electrodes. The four-layered SOC model consists of two polymethyl methacrylate sheets, a PS sheet, and a PDMS sheet. The apical chamber housed an epidermal monolayer of KC cultured on the dividing porous membrane, and the basal chamber was seeded with immune cells (U937 cell line). The immune assay chamber serves for conducting magnetic bead-based sandwiched immune assays for IL-6 and IL-8 detection. The Ag/AgCl electrodes, stationed above and below the epithelial-immune skin model, measured TEER to monitor tight junction integrity and cell monolayer confluence during cell culture. PDMS, polydimethylsiloxane; SOC, skin-on-a-chip. Reprinted with permission from Royal Society of Chemistry. 58
Wufuer et al. mimicked inflammation and edema on a chip. 59 Each polydimethylsiloxane (PDMS) layer of this model contained a representative cell type: HaCaT for the epidermis, human foreskin FB line (HS27) for the dermis, and EC for the vascular component. The three PDMS membranes were separated by porous PET membranes, which ensured nutrients and oxygen diffusion across the layers. Inflammation was induced by perfusing the microfluidic system with tumor necrosis factor alpha (TNF-α). Following TNF-α exposure, high proinflammatory cytokine levels were reported. Upon treatment with dexamethasone, a drug that reduces inflammation, tight junction analysis, and permeability testing showed reduced inflammation and edema. The authors indicate that their model was the first of its kind to mimic the healing of a pathological condition using a small-molecule drug. Although some quarters hold that this system mimics native physiology, in our opinion, it deviates from the physiological structure of skin in vivo, whereby direct dermo–epidermal interactions are key to skin homeostasis. 60 Furthermore, including an immune cell representative, rather than TNF-α, is worth investigating and is perhaps more physiologically relevant for DFU modeling.
Mori et al. developed a vascularized SOC model to study the effect of vascular perfusion on percutaneous absorption. 61 Sacrificial nylon wires were used to create hollow channels within the dermal compartment (consisting of FB-embedded collagen matrices), which were subsequently coated with EC and perfused with culture medium. The epidermal layer, consisting of human epithelial KC seeded onto a silicone rubber barrier, overlaid the reconstructed vascularized dermis. A native dermo–epidermal morphology and barrier function were both confirmed. This vascularized SOC model looks promising for studying immune cell circulation and entry into the skin tissue, and in particular, how such events can affect wound healing.
Biglari et al. introduced a dermal wound-on-a-chip with both vascular and immune components. 62 The developed SOC permits cell–cell interactions under inflammatory conditions. Monolayers of human dermal FB and macrophages were separately cultured on laterally displaced microchannels, and human umbilical vein endothelial cells (HUVEC) were cultured in Matrigel in the central one (Fig. 3a, b). This wound model mimicked the inflammatory phase of wound healing and, in the authors' opinion, can be used for testing anti-inflammatory drugs. In our opinion, including an epidermal representation to improve/utilize this layout for DFU modeling will help understanding how the vascular and immune components can contribute to reepithelization –using diseased cells.
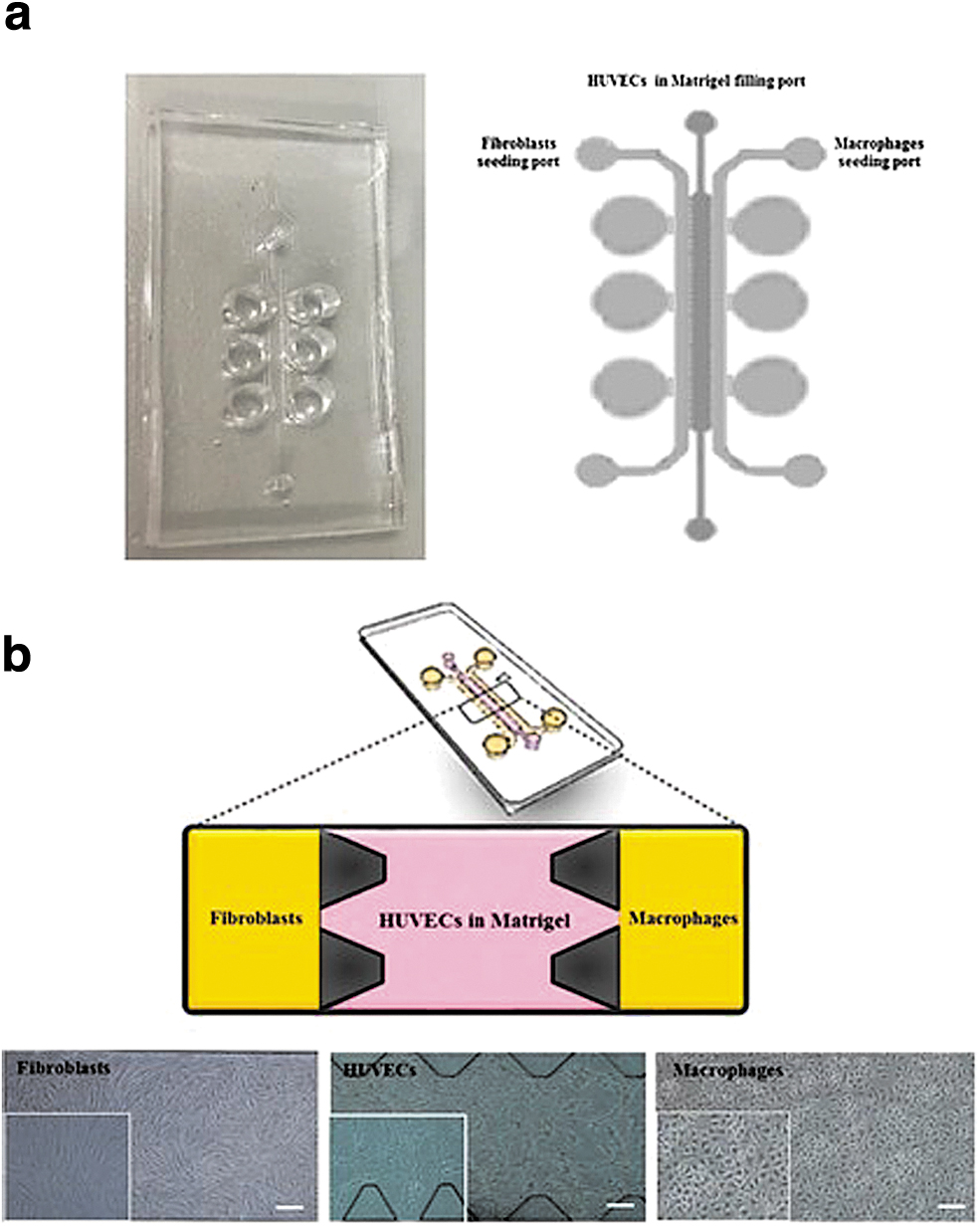
Illustration of the fabricated human wound-on-chip PDMS-based model.
Jeon et al. developed a SOC consisting of two PDMS layers separated by a 0.4 μm porous membrane, perfused by a gravity flow system, for studying hyperkeratosis, a side effect of the chemotherapeutic drug sorafenib. 63 The HSE consisted of human FB embedded within rat tail collagen (RTC) type I hydrogel, overlaid with human KC. Similarities between clinical manifestations in patients following sorafenib administration and this model were observed.
Most recently, Kwak et al. indeed progressed the field of creating immunocompetent SOCs. 64 Uniquely, their model mimicked immune cell recruitment and transport through the vascular endothelium to the site of inflammation. The top PDMS layer housed the dermal and epidermal components of the HSE (human dermal primary FB and HaCaT/primary KC, respectively); while the lower PDMS layer had a fluidic channel, with HUVEC cultured under the porous membrane that interfaced the two PDMS layers (Fig. 4a, b). The chip was perfused using a gravity-driven system (Fig. 4c). Following UV exposure of the HSE, human leukemia-60 line (HL-60) cells were added; a significantly increased migration of HL-60 leukocytes to the dermal layer was observed, compared to the control where no UV irradiation was used (Fig. 5a–d). This proves that this model is capable of responding to external stimuli as native skin. This model can be developed/adopted for studying immune-related skin conditions like DFU and for testing skin sensitizers/irritants. However, using human cell lines to represent the immune component may not be physiologically robust, given their genetic/phenotypic shift tendency compared to native cells.
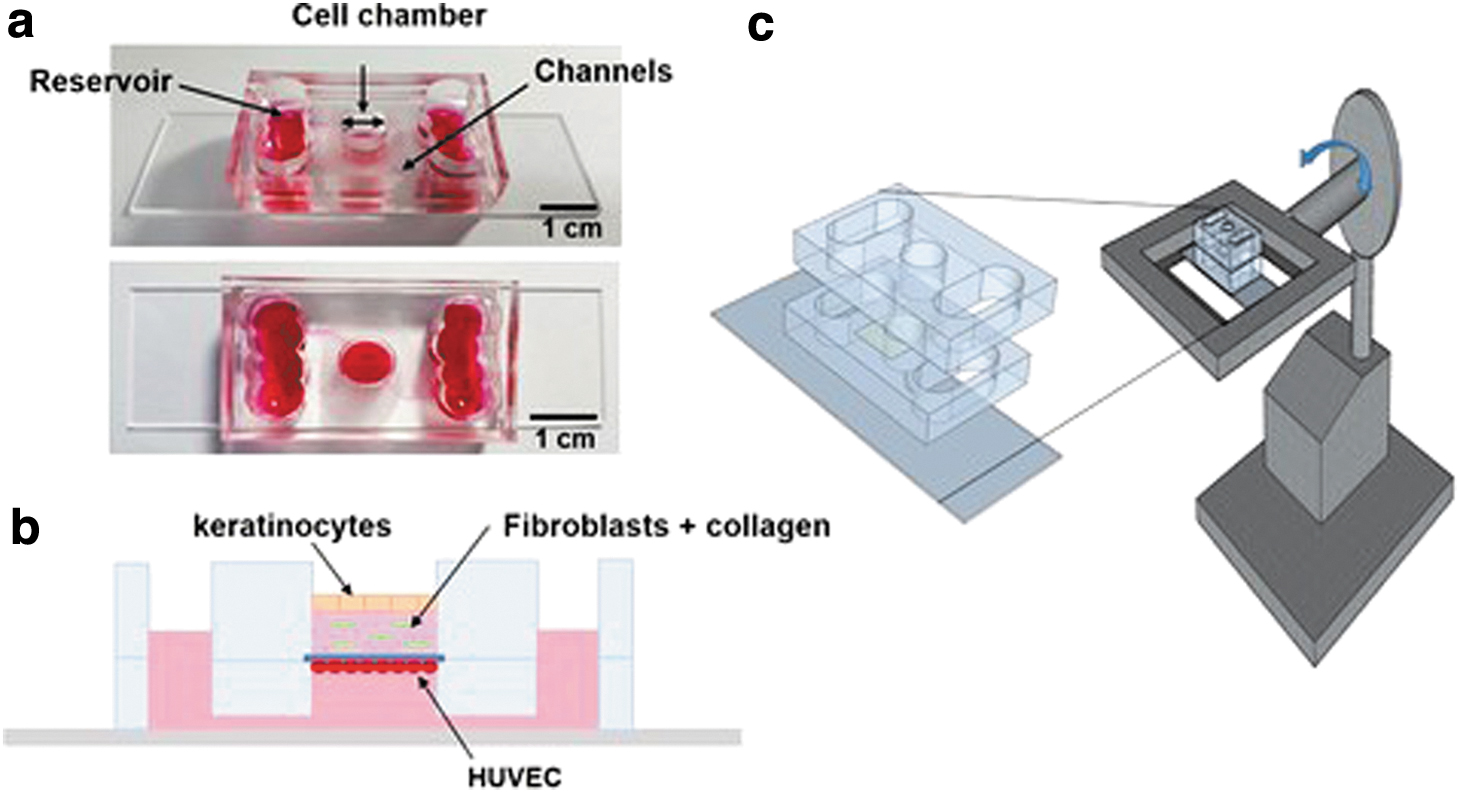
A microfluidic skin chip with vasculature that recapitulates the immune response of the skin tissue.

Inflammatory response of the immunocompetent microfluidic skin chip with vasculature following UV irradiation
Overall, these models are reminiscent in modeling DFU using microfluidics. An additional feature worth including is simulating immune cell migration and extravasation from the blood to injured site in response to cytokine/chemokine gradients. For example, Han et al. developed a blood vessel-on-a-chip to mimic neutrophil extravasation in response to chemokine gradients (Fig. 6). 65
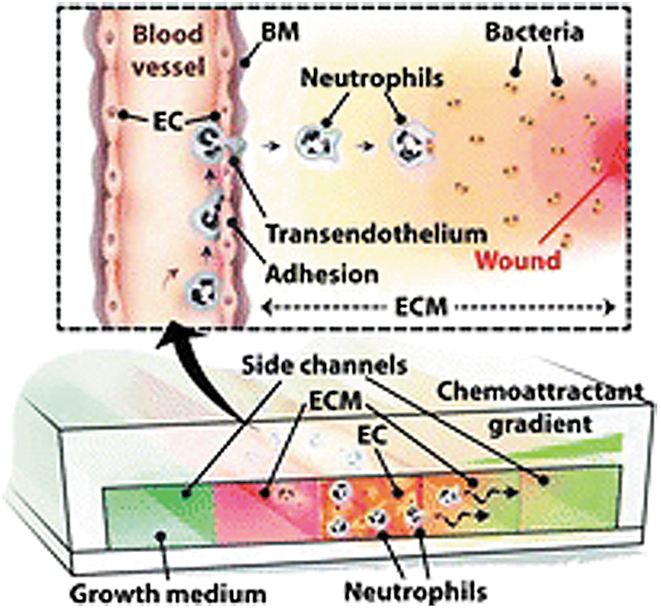
A blood vessel-on-a-chip used to model neutrophil extravasation in response to chemoattractant gradients. In vivo, neutrophils from the peripheral blood are attracted to the wound site by cytokine/chemokine gradients. N-formyl-methionyl-leucyl-phenylalanine and IL-8 were used separately for chemokine gradient generation in the fabricated blood vessel-on-a-chip model. The quantity and migratory profile of neutrophils that traversed both the ECM layer and the EC monolayer were measured. Reprinted with permission from Royal Society of Chemistry. 65
In the next section, we will consider the essential components in modeling DFU using organ-on-a-chip (OoC) technology, which is to be developed yet.
Considerations for modeling DFU using OoC technology
Microfluidics
Some limitations of conventional skin bioengineering approaches for modeling DFU can be resolved using the emerging OoC technology. OoC does not consist of constructing a whole tissue or organ in vitro, rather, it recreates a minimally functional unit of human physiology in a direct manner. 66 It is the combination of microfluidics and 3D cell culture to obtain tissues/organs on a microscale. OoC technology can significantly reduce clinical trial failure rates by considering differences in genetics, gender, and ethnicity, among other factors. 67
Microfluidics is the science of manipulating of 10−9–10−18L of fluids within microfabricated channels with at least one dimension less than 1 mm.68,69 Microfluidics provides an opportunity to fabricate the microscale in vivo environment, precisely controlling both the structure and flow. 70 Consequently, less reagent quantities are consumed, resulting more cost-effective for already developed microfluidic systems. For example, Abaci et al.'s study showed that the volume of culture medium and cell numbers used in their SOC platforms are reduced by 36-fold compared to the conventional Transwell insert systems. 71
Nutrients, biomechanical, and biochemical cues can be introduced in a controlled manner to mimic native tissues using microfluidics.72,73 Microfluidics introduces shear stress, which, when precise, facilitates cell viability and function unlike static tissue culture systems. 74 This feature is helpful for achieving longer culturing periods while maintaining tissue integrity, especially for studying side effects of drugs that are slow to manifest in dose–response studies. 67 Furthermore, epidermal differentiation and morphogenesis, and barrier function are significantly improved under controlled ventilation and continuous perfused conditions. 75 Finally, blood flow is essential for systemic transport of nutrients and immune cells within the dermal layer of the skin. 76 Hence, microfluidics will permit the recreation of circulation and immune cell movement in and out of physiological-scale engineered tissues. The disadvantages of microfluidics for OoC purposes include the associated high development costs, the key expertise needed, and technical challenges such as air bubble formation in perfused microchannels, which increases wall shear stress that in turn affects cell viability adversely.77,78
Among polymers used for microfluidic fabrication for SOC modeling, PDMS is preferred due to its low cost, minimal cytotoxicity, ease of processing, cyclic stretching potential, and high gas permeability, which is ideal for oxygen entry to cells within microsystems. PDMS also has reasonable optical transparency, which is good for real-time read-out of cell morphology and migration.79,80 However, alternative substrates (e.g., polystyrene [PS], polycarbonate, and polysulfone) 81 or surface engineering approaches are being considered to overcome hydrophobic molecule absorption, which renders PDMS unsuitable for pharmaceutical analysis.80,82–84
Soft lithography is the most common microfabrication approach for microfluidics. 85 Although popular, soft lithography has limits in its applicability for mass production. Sriram et al. developed a perfusable skin model with enhanced physiology and barrier function using microstructured polymethyl methacrylate, which the authors deemed more suitable for mass production and high throughput. 75 Another proposed microfabrication approach, although not popular but worth refining, is the use of laser ablation technologies for reconfigurable microchannel fabrication.86,87
OoC has not yet been explored for modeling DFU. Based on the literature, nonhealing DFU are strongly defined by chronic and stagnant inflammation: preventing the critical transition from the inflammation phase to the proliferation one, necessary for healing. Hence, it is worth-investigating the role of key immune cells in relationship to why DFU are hard-to-heal and their response to therapeutics using OoC technology.
Scaffold choice
Collagen is the most abundant protein of the skin's native ECM 88 and presents adhesion sites for cells.89,90 Hence, it has been extensively used for representing the dermal compartment by embedding with FB in SOC models. RTC, which supports skin formation, is particularly popular.63,73,91 However, as KC, FB and macrophages secrete collagenases, collagen undergoes cell-mediated contraction in vitro. This renders its use as nonideal for long-term cell culture due to the reduced structural integrity.92–94 Mechanical strength required to maintain the structural integrity can be improved either by chemical modification, dehydration, copolymerization, or using perfusion conditions.52,95 Collagen contraction may be overcome also by using alternative “stronger” hydrogels (e.g., fibrin, which showed noncontractility and improved mechanical strength, unlike the conventional collagen matrices, hence rendering certain downstream analyses feasible for drug testing and toxicology studies). 75 Nonetheless, given the animal source of RTC, which is inconsistent with the 3R's approach we advocate, it imparts cross-species variation in conducted experiments when using human tissues. Furthermore, it causes batch-to-batch variability in experiments; rendering its use nonideal for drug testing models. 96
Another solution could be the use of synthetic peptides that become swollen hydrogel systems when hydrated. Commercially available synthetic peptides (e.g., hydrogels produced by Manchester Biogel and Biogelx) are used for 3D cell culture and bioprinting applications. These hydrogels can be functionalized with ECM proteins, for example, collagen, fibronectin, and so on. These hydrogels are mechanically tunable to mimic the stiffness of the desired tissue: by simply varying peptide concentration. These hydrogels provide reproducibility, which is needed for developing reliable OoC models for drug testing.
In DFU, the characteristic poorly organized ECM contributes to the nonhealing status. 97 Due to the hyperglycemic microenvironment in diabetes, the ECM contains AGE, which are protein-sugar complexes formed by nonenzymatic reactions. 98 AGE induce the production of high TNF-α levels by macrophages, prolonging inflammation, and therefore impeding reepithelization.99,100 AGE, furthermore, cause altered cell-ECM interactions and cell behavior, leading to cell arrest and apoptosis, and decreased cell migration (due to deficient integrin binding). 101 The glycated ECM becomes resistant to protease activity due to reduced solubility and, hence, cannot be reorganized during wound repair. Maione et al.46,47 reported that DFU-derived FB deposited thinner ECM tissues with high fibronectin levels compared to their healthy counterparts. Furthermore, DFU-derived FB responded atypically in the presence of Transforming Growth Factor beta, which is known to regulate ECM production and reorganization during wound healing. Collectively, these findings signify that the ECM profile differs clearly between healthy and DFU tissues. Therefore, the use of synthetic peptides to mimic the “diseased” ECM of DFU would not fully capture the in vivo complexity of the ECM of diabetic ulcers, although attributing reproducibility in experiments. 102
Hence, we may need to consider alternatives to exogenous hydrogels for ECM recapitulation. In particular, it is useful to consider how DFU-derived FB can serve as a source for endogenous ECM deposition. The rationale is that ECM synthesized by DFU cells would possess the diseased signature similar to in vivo conditions. To stimulate the ECM deposition in vitro, macromolecular crowding (MMC) is advisable and has been previously applied in skin research 47 : by including ECM-inducing agents into culture media such as ascorbic acid, carrageenan, and so on.103,104 Without MMC, several weeks are required to achieve ECM deposition in vitro.
Another approach for endogenous ECM deposition is the generation of self-assembled dermal layers using scaffolds, for example, Alvatex® scaffolds (REPROCELL). These latter are highly porous and PS-based. When seeded with cells, the porous PS scaffold can form 3D-like structures. They provide support for endogenous ECM deposition for 3D cultures; changes in ECM can be observed in response to a drug; and de novo basement membrane formation occurs. Alvatex scaffolds have been used for in vitro skin equivalent generation. However, the culture period of the dermal equivalent to ensure enough ECM deposition that can prevent KC infiltration has to be optimized—to eventually ensure an organized, stratified, and differentiated epidermis. For example, Costello et al. 105 cultured senescent FB within Alvatex scaffolds, overlaid with adult KC, to generate an aging skin model. They found age-related changes, including reduced ECM synthesis. Roger et al. 106 bioengineered full-thickness HSE with barrier function properties using Alvatex scaffolds, commercially available cells, and defined low-serum media, which resulted physiologically similar to native human skin. The authors proposed that Alvatex scaffolds can be used to incorporate immune cells and melanocytes to simulate inflammatory response. These examples encourage the adoption of such approaches for modeling DFU with a “diseased” ECM, ensuring both reproducibility and physiological relevance.
Cell sources
The final aspect to consider is the cell sources. Immortalized cell lines are easy to access and expand in vitro. However, DFU-related cell lines are not currently commercially available, but can be developed in-house. For example, Caley et al. 107 developed and characterized a chronic skin wound FB cell line (using human telomerase) from VLU patients, and proposed it as an alternative for animal studies to understand VLU etiology and for drug prescreening. The genetic modification left intact the known VLU-specific phenotype and genotype, and impaired proliferative capacity and wound repopulation.
Using primary cells for DFU modeling would be ideal to attain the desired tissue function. They are isolated directly from skin biopsies (for FB and KC) and blood samples (in the case of immune cells sourcing). 108 For example, in Maione et al.'s publications,46,47 tissues were obtained from DFU patients via routine surgical procedures such as debridement, arthroplasty, bunionectomy and amputation. Three-dimensional cell culture studies involving primary cells from DFU show that they recapitulate in vivo conditions. For example, DFU-derived FB have altered communication with KC resulting in impaired reepithelization in vitro.44,46,47
Selection and quality control of sourced cell types, protocol optimization for cell isolation and medium composition, use of low-passage number, appropriate scaffold/hydrogel choice, ratio of different cell types, and culturing time to avoid aberrant signaling patterns are highly recommended when using primary cells for DFU modeling using OoC technology. 88 The main challenges, however, in using primary cells include their limited availability and interdonor variation. To tackle primary tissue availability issues, the setup of biobanks is advised, especially in academia.
Induced pluripotent stem cells (iPSC) are emerging as a potential cell source for HSE biofabrication109–113 and disease modeling.114,115 iPSC are adult somatic cells that are reprogrammed to their pluripotent state either (i) by inducing the expression of the transcription factors Oct4/Sox2/c-Myc/KLF4 using retroviral or lentiviral vectors 116 or (ii) using genomic integration-free methods such as episomal vectors and Sendai virus.117,118 An iPSC can self-renew indeterminately and potentially differentiate into any cell type of the three germ layers.119–122 However, not all types of cell lineages can actually be derived using iPSC technology. There is also a lack of robust protocols. A loss of epigenetic markers due to continual passaging and immaturity of derived cells can occur. Furthermore, epigenetic differences are suspected in iPSC, which could significantly impact DFU-on-a-chip models.67,123
Martin et al. 124 strongly proposed the use of iPSC to model DFU healing in vitro (Fig. 7): different cell types can be obtained from iPSC, derived from either KC or FB from diabetic patients, and then used to develop 3D organotypic skin models for wound healing studies and drug testing. Contrarily, Kashpur et al.125,126 reported how FB differentiated from iPSC (reprogrammed from primary FB from DFU patients) show promigratory attributes that could potentially heal recalcitrant diabetic ulcers. This leads to an open question: Could this signify that reengineering DFU-derived cells to their pluripotent state “heals” the cells and renders them irrelevant for disease modeling according to Kashpur et al's findings? Supposing DFU-derived iPSC had therapeutic effects, genome editing can be explored to engineer cells with DFU epigenetic traits.

Modeling diabetic wound repair using iPSC technology. Primary FB and KC, isolated from the dermis and epidermis of diabetic/nondiabetic individuals, respectively, can be reprogrammed to pluripotency using either transcription factor delivery via viral vectors or genomic integration-free methods. The obtained iPSCs are then differentiated to obtain different cell types for modeling DFU in vitro for 3D wound healing studies and drug discovery purposes. DFU, diabetic foot ulcer; iPSC, induced pluripotent stem cell. Reprinted with permission from Portland Press Ltd. 124
Conclusion
In the near future, bioengineering minimally functional DFU-on-a-chip is anticipated to be realized by addressing two aspects. First, by using the appropriate cell types that are consequential to wound repair, especially immune cells and, second, by their integration in a physiologically relevant reproducible scaffold that permits endogenous “diseased” ECM generation. Further benefits from miniaturization and control can be obtained by using a microfluidic platform. These DFU-on-a-chip models can then be used for routine testing of topical pharmaceutics as well as other regenerative therapies.
In this article, we propose the use of diseased cells, especially KC, FB, and macrophages, in reproducible scaffolds for endogenous ECM generation for DFU modeling. Key publications have been identified for these two aspects. While the progress on the cell and scaffold aspects is convincing, the integration on a microfluidic platform, however, is less well-advanced and needs further development.
For high-throughput, reproducibility, and customizable purposes, printing processes can be used for the creation of the DFU tissue and microfluidic structures. Furthermore, integrated real-time and noninvasive sensors for monitoring reliable DFU clinical biomarkers in DFU-on-a-chip models for drug testing will help with the robust data required for easing the burden imposed by chronic wounds in the future.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the EPSRC and SFI Centre for Doctoral Training in Engineered Tissues for Discovery, Industry and Medicine, Grant Number EP/S02347X/1.
G.O.’C., G.G.’s, and Y.R.'s contribution is supported by EU INTERREG project EAPA 384 2016 “AtlanticKETMed.”