
Abstract
There is critical unmet need for new vascularized tissues to support or replace injured tissues and organs. Various synthetic and natural materials were already established for use of two-dimensional (2D) and three-dimensional (3D) in vitro neovascularization assays, however, they still cannot mimic the complex functions of the sum of the extracellular matrix (ECM) in native intact tissue. Currently, this issue is only addressed by artificial products such as Matrigel™, which comprises a complex mixture of ECM proteins, extracted from animal tumor tissue. Despite its outstanding bioactivity, the isolation from tumor tissue hinders its translation into clinical applications. Since nonhuman ECM proteins may cause immune reactions, as are frequently observed in clinical trials, human ECM proteins represent the best option when aiming for clinical applications. Here, we describe an effective method of isolating a human placenta substrate (hpS) that induces the spontaneous formation of an interconnected network of green fluorescence-labeled human umbilical vein endothelial cells (gfpHUVECs) in vitro. The substrate was biochemically characterized by using a combination of bicinchoninic acid (BCA) assay, DNA, and glycosaminoglycan (GAG) content assays, sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) analysis and Western blot, angiogenesis arrays, chromatographic thrombin detection, high performance liquid chromatography (HPLC)-based amino acid quantification analysis, and assessment of antimicrobial properties. 2D in vitro cell culture experiments have been performed to determine the vasculogenic potential of hpS, which demonstrated that cell networks developed on hpS show a significantly higher degree of complexity (number of tubules/junctions; total/mean tube length) when compared with Matrigel. As 3D cell culture techniques represent a more accurate representation of the in vivo condition, the substrate was 3D solidified using various natural polymers. 3D in vitro vasculogenesis assays have been performed by seeding gfpHUVECs in an hpS-fibrinogen clot. In conclusion, hpS provides a potent human/material-based alternative to xenogenic-material-based biomaterials for vascularization strategies in tissue engineering.
Impact statement
There is a critical need for new vascularized tissues to support or replace injured tissues or organs on the one side, and a rising voice on the other side to replace animal materials in science. Here, we describe an effective method of isolating a human placenta substrate (hpS) comprising extracellular matrix proteins and bioactive factors, which provides a potent human/material-based alternative to xenogenic-material-based biomaterials such as Matrigel™ for vascularization strategies in tissue engineering.
Introduction
Over 500 million people worldwide would currently benefit from pro- or antiangiogenesis treatments. 1 Numerous pathological entities or surgical inventions could benefit from therapeutic stimulation of new blood vessel formation. 2 Wound healing, myocardial ischemia, plastic surgery, or cancer research are just a few of the many situations that could be improved through a new or regenerated blood vessel system.2,3
Hence, the success of many current therapies in regenerative medicine requires the ability to create stable, hierarchically organized vascular networks within the engineered or regenerated tissues.4,5 In any tissue or scaffold of relevant size, viable cells need to be within a distance of maximal 200 μm of preexisting blood vessels (the diffusion limit of oxygen and nutrients within tissues) to stay alive.5,53 Therefore, therapeutic stimulation of new blood vessel formation (neovascularization) is a key objective of research in tissue engineering and regenerative medicine (TERM). 67
Currently, there is a broad variety of choices when selecting scaffold biomaterials for TERM. 7 Various synthetic or natural polymers were already tested as scaffold materials for three-dimensional (3D) in vitro vasculogenesis and angiogenesis research. However, the success rate of complete vessel maturation and therefore the clinical relevance of most of these biomaterials are limited and associated with various bottlenecks. Generally, most models contain few polymers, for example, polyethylene-glycol (PEG) or collagen-I [COL1]), one distinct cell type (e.g., human umbilical vein endothelial cells [HUVECs]), and one bioactive component, for example, vascular endothelial growth factor (VEGF).
Therefore, these one-component models often reflect distinct effects in the cascade of neovascularization, but as they do not adequately mimic the natural diversity of native tissue, 8 they do not successfully induce vessel maturation, which is, however, essential for vascularized biomaterials planned for transplantation. For instance, synthetic polymers are generally cheap, well defined, and highly processable, however, they are inert and the vast majority of synthetic polymers do not exhibit cell-interactive properties. 13
In contrast, the heterogenic mixture of extracellular matrix (ECM) proteins, processed from tissues, are the most natural scaffolds.7,9,10 In nature, neovascularization is orchestrated by different molecular mechanisms of different kinds of proteins within the ECM, in total composed of over 300 different proteins, proteoglycans, and signaling molecules in humans. 11 The ECM is nature's own multifunctional scaffold,12,13 and thus, the ideal environment for human cells is provided by the human natural ECM.3,9,12,14
The ECM has a profound impact on the behavior of all eukaryotic cells,8,10 acts as the reservoir for growth factors, and exerts fundamental control over angiogenesis in all the neovascularization stages.3,14 ECM modulates a wide range of fundamental mechanisms in the development, function, and homeostasis of all eukaryotic cells.10,12 Therefore, biomaterials extracted from naturally occurring ECM have received significant attention in TERM.13,15,99–101
A prominent example is Matrigel™, a heterogenous substrate extracted from tissues derived from Engelbreth-Holm-Swarm (EHS) tumor in mouse models, which represents the gold standard for many in vitro vasculogenesis15–19,30,31 and in vivo angiogenesis20–27 studies in research.
However, the major drawback of Matrigel is that it is not intended for clinics, due to its xenogenic tumorigenic origin. 68 In addition, production of Matrigel requires the sacrifice of large numbers of animals. 17
Furthermore, many xenogenic biomaterials are still associated with immunological responses in up to 5% of all patients, 32 and harbor the risk of xenogenic pathogen contamination and potential disease transmission.33–37 Thus, their use in large clinical studies is controversially debated. In addition, many xenogenic proteins are known to have a lower clinical performance when compared with human proteins.38–40
As a consequence, human-tissue extracted ECM is regarded as the best option for the creation of new medicinal products, because the ECM structures of donors and recipients are almost identical among species. Human placenta, a medical waste product in consistent quantity and quality, is described as a tissue with a strong proangiogenic potential.41,42,44
Placenta ECM proteins are free of any ethical conflicts. 43 Placenta is globally and consistently available after birth for processing on large scales. This unique temporally human tissue harbors high amounts of various proangiogenic proteins.45,60,69 Various placenta-ECM-derived biomaterials have already been used as a biomaterial for in vitro and in vivo vasculogenesis and angiogenesis studies,43–46,72 and already integrated in routine clinical use.47,48 Placenta tissue is also reported to have very good antibacterial, anti-inflammatory, and antiscarring properties. 51
Many human placenta ECM-extracted substrates such as Placentrex® (M/s. Albert David), 47 Laennec® (Japan Bioproducts Industry), or Melsmon Cell Revitalization Extract® (Melsmon Pharmaceuticals),70,71 which are mainly extracted by use of heat and pressure, have been successfully used for decades as a topical or injectable agent in clinical approaches related to wound healing,53,67,73–75 burn injuries, postsurgical dressings, and bedsores,47,75 but their potential for neovascularization in tissue engineering is at least to our knowledge unknown. Probably, because Placentrex, for instance, contains only fragments of fibronectin and some smaller peptides, glycosaminoglycans (GAGs), lipids, and polynucleotides, but it is not highlighted to contain any active proangiogenic factors that might have survived the heat extraction.47,73,76,77
In addition, allogenic transplantation of the human amnion (hAM) for clinical applications has already been successfully performed for over 100 years.49,50 Nowadays, it is also used for ophthalmology, wound healing, and regenerative medicine purposes.48,51–55 In all these clinical studies, applications of placenta ECM components have been proven to be safe to patients.
The aim of this study was to establish an effective method of isolating a fully human placenta substrate (hpS). For the extraction, a Tris 0.5 M NaCl buffer was used and compared with substrates from the same donor, which were extracted with a Tris 2 M urea buffer, the same detergent used for the extraction of Matrigel. The resulting hpS was biochemically characterized by determining residual DNA and GAG content, identification of proteins by sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) combined with Western blot analysis, and total amino acid quantification. Chromogenic thrombin detection techniques and assessment of the angiogenic profile using angiogenesis array and antimicrobial properties of hpS were further assessed.
In two-dimensional (2D) in vitro assays, the influence of the cell number and the variation between single donations for in vitro cell network formation (vasculogenesis) was assessed using green fluorescence-labeled human umbilical vein endothelial cell (gfpHUVEC) or NIH3T3 fibroblasts. As the prevalence of 3D in vitro systems is increasing due to the ability to mimic the in vivo environment, hpS was solidified by mixing it with various natural polymers. For the 3D in vitro experiments, hpS was mixed with clinically used fibrinogen.
Methods
If not stated otherwise all reagents were obtained from Sigma Aldrich and are of analytical grade.
Collection of human placenta tissue
Placenta material was provided by the tissue bank of the Red Cross Blood Transfusion Service of Upper Austria in Linz and collected after cesarian section at the Kepler University Clinics Linz, Austria (with the consent of the local ethics board and informed consent from all donors). Tissues were stored at −20°C up to 3 months until isolation of an hpS was performed.
Biochemical characterization of hpS
Total protein content
Protein content of hpS was determined using a bicinchoninic acid (BCA) assay (23228; Thermo Scientific, Vienna, Austria), according to the manufacturer's instructions. Briefly, dilutions of bovine serum albumin (BSA) were used to generate a standard curve. Samples/standards and BCA buffer were pipetted into 96-well plates (Greiner Bio-One, Kremsmünster, Austria) and incubated at 37°C for 30 min. Then, the absorbance was measured at 562 nm using an Omega POLARstar 140 plate reader (BMG Labtech, Ortenberg, Germany).
Papain digestion
Papain digestion was performed as described elsewhere.43,56 Freeze-dried hpS was digested with 3 IU/mL papain from papaya latex (75 mM NaCl, 27 mM Na citrate, 0.1 M NaH2PO4, 15 mM EDTA, and 20 mM
DNA content
CyQUANT stain was used as described by the manufacturer for DNA quantification. Briefly, papain-digested samples and standards from DNA sodium salt from calf thymus were pipetted into 96-well black microplates (Brand, Wertheim, Germany). The plate was incubated in the dark for 5 min at room temperature. Then, the fluorescence intensity was measured using an Omega POLARstar 140 plate reader (BMG Labtech) at 485 nm with a reference wavelength of 520 nm.
GAG quantification
Dimethyl methylene blue (DMB) was used as described for GAG quantification.43,57 Papain-digested samples were diluted in Aqua Dest before measurement and 100 μL of standard/samples was pipetted into 96-well plates (Greiner Bio-One flat bottom). Two hundred microliters of DMB color solution (46 μM DMB, 40 mM NaCl, 40 mM Glycine in dH2O, pH 3) was added and optical absorbance was immediately measured at 530 nm with a reference wavelength of 590 nm using an Omega POLARstar 140 plate reader (BMG Labtech).
SDS-PAGE gel electrophoresis/Western blot
SDS-PAGE and Western blot analysis were performed as described for protein specification58,59 using the XCell SureLock™ Mini-Cell Electrophoresis System (Invitrogen, Vienna, Austria). Twenty micrograms of sample proteins per lane was resolved on 3–8% gels and a marker (Gel filtration standard 151-1901; BioRad, Vienna, Austria) and 12% gels and a marker (Protein marker V; VWR, Vienna, Austria). The gels were stained with Coomassie Brilliant Blue R-250 (BioRad), or transferred onto nitrocellulose membranes for Western blot analysis (Peqlab).
The membranes were blocked with 5% milk in TBS buffer containing 0.1% Tween (TBS/T), and primary antibodies against COL1 (AB 34710; Abcam, Cambridge, MA), collagen-IV (AB6586, Abcam), or laminin-111 (AB11575, Abcam) in 5% BSA-TBS/T were incubated at 4°C overnight. Membranes were further incubated in 5% milk-TBS/T for 1 h containing secondary antibodies (LI-COR Biosciences, Lincoln, NE) and the signals were detected using the Odyssey Fc infrared imaging system (LI-COR Biosciences).
Angiogenesis array
Relative levels of angiogenesis-related proteins from hpS Tris-urea or Tris-NaCl were determined using human angiogenesis antibody arrays C1000 (RayBio) according to the manufacturer's instructions.45,60 Membranes containing 43 different cytokine antibodies (duplicates) were blocked and incubated with 1 mL of three pooled, normalized hpS samples overnight at 4°C. All residual steps were performed at room temperature. After washing, biotinylated antibody incubation for 2 h, and a second wash, the membranes were incubated with HRP streptavidin for 2 h, washed, and chemiluminescence was detected using myECL Imager (Thermo Scientific, USA).
Data analysis was performed according to the manufacturer's instructions. Each membrane was exposed to obtain high signal-to-noise ratios using the gel documentation system (myECL Imager; Thermo Scientific, USA). The spot signal intensities were further analyzed using myImageAnalysis Software Version 1.0 (Thermo Scientific, USA). One array was defined as “Reference Array,” to which the other arrays were normalized to and a working template was created. For each spot, the signal density (intensity/area) was used for numerical data transformation. The background signal was subtracted from raw numerical densitometry data and normalized to the positive control signals—standardized amounts of biotinylated IgG.
Chromogenic thrombin assessment
Human thrombin was assessed using chromogenic measurements (Technothrombin®TRA; Technoclone, Vienna, Austria) according to the manufacturer's instructions. Briefly, the detergents were diluted in Aqua Dest and pipetted in black NUNC 96-well plates, and calibration curves were measured at 37°C using a fluorometer (BMG Labtech) at 360/460 nm extinction/emission for 10 min in 30-s measurement intervals, before the analysis of hpS Tris-NaCl was assessed for 60 min in 1-min measurement intervals. All plate readings were immediately performed after pipetting the samples/substrate.
Characterization of antimicrobial effects of hpS Tris-NaCl
hpS Tris-NaCl from three different isolations was pooled and ultraviolet (UV) sterilized in six-well plates for 30 min. Aliquots were stored at −20°C until further use. The bacterial strains (Table 1) were grown in Lysogeny broth (LB medium; LB Broth, Molecular Genetics Granular; Miller) o/n without antibiotics. Then, the cultures were diluted 1:6 to 1:10 with fresh medium and grown for 30 min with shaking (200 rpm) at 37°C to exponential growth phase (optical density [OD]600 0.5–0.7). Based on the OD600 measurement, the bacterial concentrations were calculated according to the formulas given in Table 1 and the suspension was diluted to 2 × 106 bacteria/mL. Fifty microliters of these dilutions (1 × 105 bacteria) was mixed with 50 μL of hpS Tris-NaCl in a flat-bottom 96-well plate. OD600 values were measured with an Omega POLARstar 140 plate reader (BMG Labtech) for a total time of 7 h.
Bacterial Strains Used
OD, optical density.
For the negative controls, hpS Tris-NaCl was replaced by phosphate-buffered saline (PBS). Each sample was measured in triplicate, and the experiment was performed three times for statistical analysis.
Amino acid analysis
Amino acid quantification was performed as previously described, 61 using three hpS samples from three different placentas.
Sample preparation
Freeze-dried hpS Tris-NaCl was digested following a two-step protocol: first enzymatically and then chemically. Briefly, 75 mg of lyophilized sample was incubated with 1 mL of 0.0125% protease from Streptomyces griseus in 1.2% Tris/0.5% SDS pH 7.5 (adjusted with 0.1% HCl) solution for 72 h at 37°C. Then 1 mL of 4% formic acid in ddH2O was added for chemical predigestion and the suspension was incubated for 2 h at 108°C followed by lyophilization.
The dried samples were then incubated for 2 h with 5 mL of a solution containing 0.6% TRIS and 7 M guanidinium hydrochloride pH 8. After centrifugation (Sigma centrifuge, 3–18 K) of the sample at 4,800 rpm for 15 min at 4°C, 1 mL of the supernatant was combined with 0.5 mL 4 M methanesulfonic acid solution containing 0.2% tryptamine and was incubated for 1 h at 160°C. Subsequently, the solution was quantitatively transferred into a 5 mL volumetric flask, and 225 μL 8 M NaOH and 0.25 mL internal standard were added and the flask was filled up with 2.2 M sodium acetate solution. The samples could then directly be used for high performance liquid chromatography (HPLC) analysis.
HPLC standard preparation
A multiamino acid standard mixture was prepared by mixing the amino acid standard, a solution containing 2.5 mM each of asparagine, glutamine, and tryptophan, in MQ, a solution containing 2.5 mM of hydroxyproline in 0.1 M HCl and a solution of the internal standards, that is, 25 mM each of norvaline and sarcosine in 0.1 M HCl. Ten different concentrations of this standard mixture, ranging between 45 and 0.5 mg/L, were used for calibration.
HPLC analysis
The HPLC system Ultimate 3000 (Thermo Fisher Scientific, USA) was equipped with a pump (LPG-3400SD), a split-loop autosampler (WPS-3000 SplitLoop), a column compartment (TCC-3000SD), and a fluorescence detector (FLD-3400RS). Chromeleon 7.2 was used for the control of the device as well as for the quantification of the peak areas. Chromatographic separation was achieved with a reversed-phase column (Agilent Eclipse AAA, 3 × 150 mm, 3.5 μm), a guard column (Agilent Eclipse AAA, 4.6 × 12.5 mm, 5 μm), and a gradient using eluant (A) 40 mM NaH2PO4 monohydrate pH 7.8 and eluant (B) MeOH/ACN/MQ (45/45/10, v/v/v). The protocol was run with a flow rate of 1.2 mL·min−1, the column oven temperature was set to 40°C, and the injection volume was 10 μL.
As most amino acids have no fluorophore in their structure, an in-needle derivatization step was performed using 0.4 M borate buffer, 5 mg/mL ortho-phthalaldehyde (OPA) in 0.4 M borate buffer containing 1% of 3-MPA, 2.5 mg/mL FMOC, and 1 M acetic acid for pH adjustment. To guarantee sample quantification despite the derivatization step, every sample was spiked with 25 mM sarcosine in 0.1 M HCl and 25 mM norvaline in 0.1 M HCl as internal standards. Primary amines and norvaline were detected at Ex 340 nm/Em 450 nm and secondary amines and sarcosine were detected at Ex 266 nm/Em 305 nm.
Experiment
hpS isolation
All isolation steps were performed in a cold room at 4°C. After thawing, the amnion, chorion, and umbilical cord were removed. The resulting basal villous tissue was used for the isolation process (Fig. 1). Blood components were removed by repetitive homogenization steps, where 200 g of basal placenta tissue was homogenized in 400 mL of Tris NaCl buffer (0.05 M Tris, 3.4 M NaCl, 4 mM EDTA, 2 mM N-ethylmaleimide [NEM], pH 7.4) using a grinder (Braun Type 4184) and subsequently centrifuged at 7,000 g for 5 min using a Heraeus Multifuge (Beckman Instruments GmbH Type 1 S-R). The supernatant containing blood components was discarded and the pellets resuspended in 400 mL of fresh Tris-NaCl buffer. This procedure was repeated two additional times.

Flowchart for the isolation of hpS from term placenta (1). After basal tissue collection (2), the main blood components were removed by subsequent homogenization and centrifugation steps (3). Finally, hpS was isolated by salt precipitation using a Tris 0.5 M NaCl buffer (4), centrifugation (5), and PBS dialysis (6) to yield hpS. hpS, human placenta substrate; PBS, phosphate-buffered saline. Color images are available online.
For hpS extraction, 100 g of pellets were suspended in 100 mL of either a Tris-NaCl buffer (0.05 M Tris, 0.5 M NaCl, 4 mM EDTA, 2 mM NEM, pH 7.4) or a Tris-urea buffer (0.05 M Tris, 2 M urea, 0.15 M NaCl, 4 mM EDTA, 2 mM NEM, pH 7.4) and stirred for 24 h on a magnetic stir plate at 200 rpm at 4°C. The suspensions were centrifuged at 14,000 g for 20 min. The pellets were discarded (some pellets were kept for additional measurements; a second precipitation step), and the supernatants containing hpS were collected and dialyzed against 40 × volume PBS buffer in 6–8 kDa cutoff dialysis membranes (No. 21152-5; Fisher Cellulose).
PBS was changed three times. The resulting substrates (hpS Tris-NaCl; hpS Tris-urea) were stored at −80°C. Aliquots of hpS were further dialyzed against 40 x volume Aqua Dest in 6–8 kDa cutoff dialysis membranes (No. 21152-5; Fisher Cellulose) to remove the remaining salts and freeze-dried, and an amino acid quantification was performed.
3D solidification of hpS Tris NaCl
Collagen-I
Freeze-dried COL1 from human placenta 61 was resolved in PBS buffer to a concentration of 8 mg/mL, hpS was added (1 + 1 vol.), and the final solution was incubated at 37°C to achieve solidification.
Gelatin
Gelatin (4078; Merck) was diluted in hpS at room temperature to a final concentration of 3% and the solution was incubated at 4°C to achieve solidification.
Fibrinogen
Fibrinogen (Tisseel; Baxter) was diluted in the EGM-2 medium to a concentration of 10 mg/mL, only hpS was added (1 + 1 vol.), and the final solution was incubated at 37°C to achieve solidification.
Agarose
Agarose (Biozym LE Agarose, Oldendorf, Germany) was resolved in aqua dest to a concentration of 2% at 175°C until the suspension became clear. After cooling to 40°C, hpS was added (1 + 1 vol.) and the solution was incubated at 4°C to achieve solidification.
Agar-agar
Agar-agar (Fluka, St. Louis, MO) was resolved in Aqua Dest to a concentration of 3% at 90°C and after cooling to 40°C, hpS was added (1 + 1 vol.) and the solution was incubated at 4°C to achieve solidification.
2D in vitro bioactivity
HUVEC isolation
HUVECs were isolated from three donors as previously described. 62 HUVECs were isolated from umbilical cord donated with the placenta from healthy donors with informed consent and proven by the Local Ethics Committee of Upper Austria. Cells (p5–p9) were cultured in EGM-2 (Lonza, Italy), supplemented with 5% fetal calf serum (FCS). Isolated HUVECs were retrovirally infected with expression vectors for fluorescent proteins using the Phoenix Ampho system as described. 63
HUVEC seeding density
Vasculogenesis assays were performed as described.8,64,65 Briefly, 50 μL of hpS Tris-NaCl or hpS Tris-urea extracted from the same tissue was pipetted in 96-well plates, UV sterilized for 30 min, and incubated at 37°C for 3 h. Thereafter, different cell numbers ranging between 5,000 and 25,000 HUVECs from the same donor (passage 8) were seeded on hpS in 100 μL of EGM-2 medium (Lonza, Basel, Switzerland).
After 2 days of cultivation, the formed cell networks were imaged and analyzed as described. 66 Fluorescence microscopic pictures were taken from two different fields per well with a Leica epifluorescence microscope DMI6000B (Vienna, Austria) and processed in a blinded way using Adobe Photoshop software (Adobe Systems, San Jose, CA) by adjusting contrast and brightness. Then, tube formation was analyzed using AngioSys 2.0 software (TCS Cellworks, London, United Kingdom) and the AngioSys values were statistically analyzed using Prism 5 (GraphPad Software, San Diego, CA).
Immunohistochemistry
Formed HUVEC networks on hpS Tris-NaCl were stained with anti-CD 31 and vascular endothelial cadherin (VECad) antibodies (BD Pharmingen, San Diego, CA) after 2 days of cultivation. The medium was aspirated and cells were washed with PBS before fixation in 4% formaldehyde for 10 min and washing with PBS for 5 min. All following steps were performed in the dark. Cells were incubated in PBS containing 1% BSA and CD31 antibody (555445; BD Pharmingen) mouse α-human 1:100; or VECad antibodies (560411; BD Pharmingen) mouse α-hum 1:100 for 30 min at room temperature.
Then the cells were washed twice with PBS for 5 min and the secondary antibody AK Alexa Fluor 488 goat αmouse IgG (a11029, 1:100; Life Technologies) in PBS containing 1% BSA was added and incubated for 30 min at room temperature. Plates were washed twice with PBS for 5 min and DAPI was added (1:1,000). After a final PBS washing step, the networks were imaged.
Comparison of substrates
To determine the influence of substrates on the cell networks, 50 μL of Matrigel, hpS Tris-NaCl, or Tris-urea from the same tissue was pipetted in 96-well plates, UV sterilized, and incubated at 37°C for 3 h. Twenty thousand gfpHUVECs from a donor (p8) were seeded in 100 μL of EGM-2 medium (Lonza). After 3 h, the medium was replaced with 100 μL of minimally essential RPMI-1640. Medium change was performed every second day and the networks were analyzed after 6/24/48/72/96/120 h.
Single-placenta tissue comparison
To determine the consistency of the isolation method using single tissues, hpS Tris-NaCl was isolated from three different tissues, each weighing around 500 g. Fifty microliters of Matrigel or hpS was pipetted in 96-well plates, UV sterilized, and incubated at 37°C for 3 h. Twenty thousand gfpHUVECs from a donor (p7) were seeded in 100 μL of EGM-2 medium (Lonza). After 3 h, the medium was replaced with 100 μL of minimally essential RPMI-1640 medium, and the networks were analyzed every 24 h.
gfpNIH3T3 fibroblast cultivation
NIH3T3 mouse fibroblasts were purchased from DSMZ (No. ACC59, Braunschweig, Germany) and cultured in Dulbecco's modified Eagle's medium (DMEM) high-glucose supplemented with 10% FCS and 1% glutamine.
Fifty microliters of Matrigel or hpS Tris-NaCl was pipetted in 96-well plates, UV sterilized for 30 min, and incubated at 37°C for 3 h. Then, 20,000 gfpNIH3T3 fibroblasts were seeded on coated or uncoated wells (control) in 150 μL of DMEM, and after 24 h, the cells were analyzed.
HUVEC culture supplementation
To determine the potential of hpS Tris-NaCl as a cell culture medium supplement, 20,000 gfpHUVECs from a donor (p7) were seeded in 150 μL of EGM-2 medium (Lonza) or EGM-2 medium supplemented with 30% of UV-sterilized hpS in uncoated 96-well plates, or in 150 μL of EGM-2 medium on hpS 0.5 M Tris-NaCl-coated plates or on a Tris 0.15 M NaCl-extracted substrate. The networks were analyzed after 24 h.
3D in vitro bioactivity
Mixing hpS Tris-NaCl with fibrinogen for 3D studies
hpS Tris-NaCl was pipetted in 6-well plates and the wells were UV sterilized for 30 min. Meanwhile, fibrinogen (Tisseel; Baxter) was diluted in EGM-2 medium to a concentration of 20 mg/mL at 37°C. Five hundred microliters of this suspension was mixed 1:1 vol. with 500 μL of EGM-2 medium containing 500,000 gfpHUVECs. This suspension was further mixed (1:1 vol.) with hpS or 0.4 U thrombin (Tisseel; Baxter) as sample control and incubated at 37°C for 2 h. After polymerization, 3 mL of EGM-2 medium was added. Medium was changed every third day and the wells were analyzed after 8 days of cultivation.
Scanning electron microscopy
Scanning electron microscopy (SEM) analysis of fibrin gels was performed as described. 61 The gels were fixed in 4% formaldehyde followed by sample dehydration using graded ethanol concentration series and hexamethyldisilazane. Samples were sputter-coated with Pd-Au using a Polaron SC7620 sputter coater (Quorum Technologies Ltd.), and examined at 15 kV using a JEOL JSM-6510 scanning electron microscope (Jeol GmbH).
Statistical analysis
All experimental data are presented as mean ± standard deviation if not stated otherwise, and p-values <0.05 were considered statistically significant. Normal distribution of data was tested with the Kolmogorov–Smirnov test before analysis using analysis of variance. All calculations were performed using GraphPad Prism version 6.00 (GraphPad Software).
Experimental Results
Extraction of hpS
A flowchart of the isolation method is depicted in Figure 1. In average, around 300–350 mL of hpS was extracted from ∼500 g of single-placenta tissues.
Compositional assessment of hpS
To assess the total protein concentration of both substrates, the BCA assay was performed (Fig. 2A). The protein concentration of hpS Tris-NaCl (1.74 ± 0.26 mg/mL) was significantly lower when compared with hpS Tris urea (2.26 ± 0.32 mg/mL).
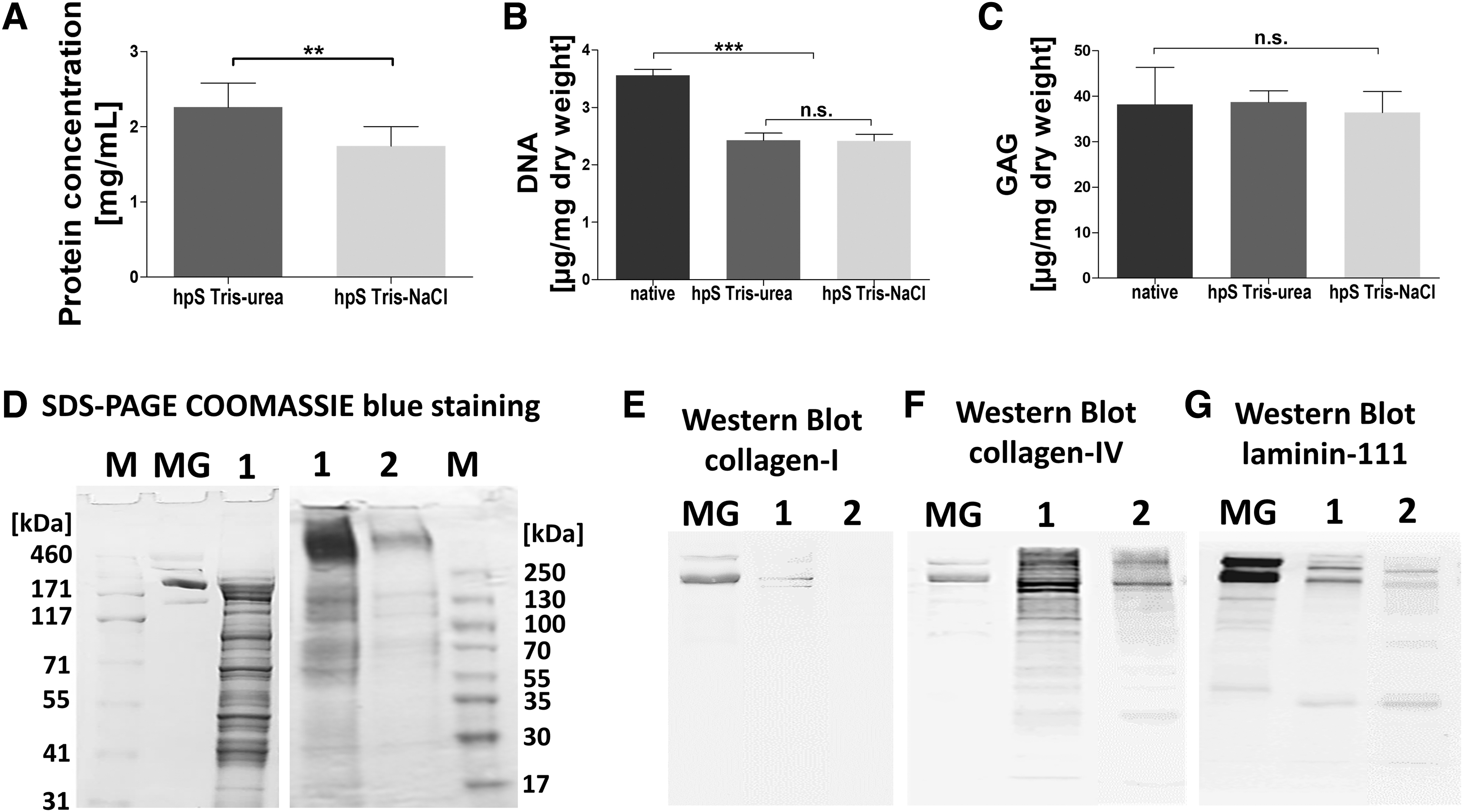
hpS contains a heterogenic mixture of proteins.
To assess the DNA content in hpS, the CyQUANT stain was used (Fig. 2B). The mean DNA content of both hpS was significantly lower compared with native placenta tissue (3.56 ± 0.10 μg/mg dry weight), but no significant difference between hpS Tris-urea and hpS Tris-NaCl was detected (hpS Tris-urea 2.42 ± 0.05 and hpS Tris-NaCl 2.41 ± 0.02 μg/mg dry weight).
DMB assays were performed to determine the GAG content within hpS (Fig. 2C). There was no significant difference among native placenta, hpS Tris-urea, and hpS Tris-NaCl (38.21 ± 6.64, 38.74 ± 2.12, and 36.4 ± 4.04 μg/mg dry weight), respectively.
SDS-PAGE was performed to visualize the composition of proteins in hpS Tris-NaCl (1 in Fig. 2D). hpS Tris-NaCl shows various protein bands ranging from 20 kDa up to around 500 kDa, whereas Matrigel consisted of fewer protein bands. A second use of the pellet (after the first Tris-NaCl precipitation) for an additionally second Tris-NaCl precipitation step yielded lower protein concentrations (2 in Fig. 2D). Western blot analysis shows that COL1 was only present in hpS Tris-urea, whereas collagen-IV and laminin-111 were detected in both hpS (Fig. 2E–G).
An antibody-based angiogenesis array was used to assess the angiogenic profile of hpS (Fig. 3). There were higher levels of in total 43 different proteolytic enzymes, immune-related cytokines, growth factors, and angiogenic chemokines assessed in hpS Tris-NaCl when compared with hpS Tris-urea. Angiogenin, a potent stimulator of angiogenesis, was the most prevalent angiogenic chemokine in both hpS.

Angiogenic profile of hpS Tris-urea and hpS Tris-NaCl in normalized intensity to the positive control, standardized IgG (n = 3).
Other chemokines including angiogenin (ANG), growth-regulated oncogene (GRO), angiopoietin or tissue inhibitors of metalloproteinases (TIMPs), proteolytic enzymes (metalloproteinases [MMP-1, MMP-9]), interleukins (IL-1 β), or cytokines related to wound healing and tissue regeneration (transforming growth factor-β1 [TGF-β1], basic fibroblast growth factor [bFGF], epidermal growth factor [EGF], platelet-derived growth factor [PDGF], and insulin-like growth factor 1 [IGF-1]) were also detected. 46
A chromogenic assay was performed to assess the presence of active thrombin in hpS Tris-NaCl. In average, 0.63 ± 0.16 U thrombin per mL was detected in hpS Tris-NaCl.
The antimicrobial effects of hpS Tris-NaCl were tested in two gram-negative strains (Escherichia coli TOP10, E. coli MG1655) and two gram-positive strains (Staphylococcus carnosus, Staphylococcus capitis). In S. carnosus, hpS Tris-NaCl showed distinct antibacterial properties and significantly delayed bacterial growth over 7 h. However, in the other strains, hpS showed a positive effect on bacterial growth (Fig. 4).
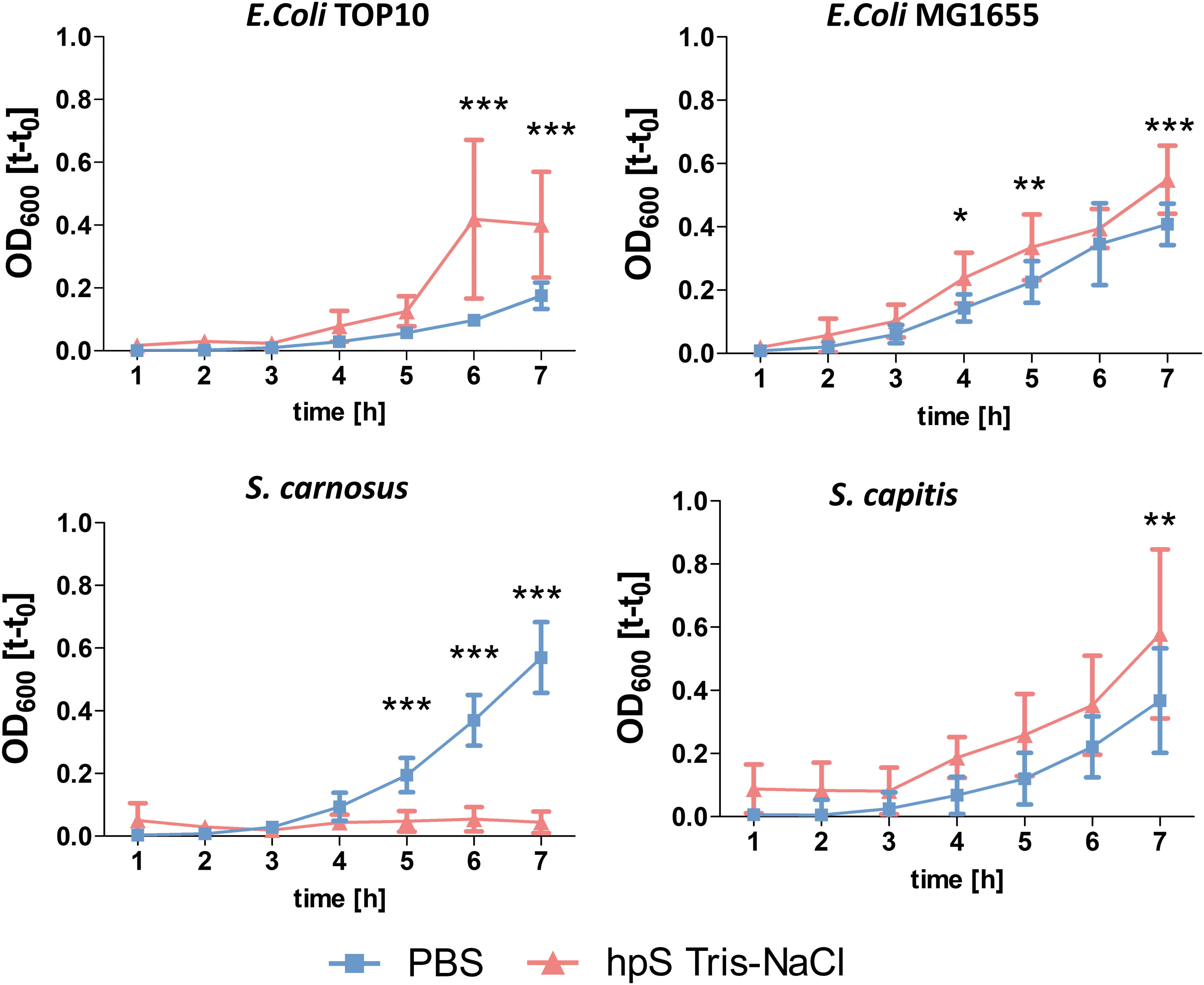
Antimicrobial properties of hpS Tris-NaCl and PBS (control) tested in two gram-positive and gram-negative bacterial strains (n = 9). *p < 0.05, **p < 0.01, and ***p < 0.001. Color images are available online.
Table 2 lists the amino acid composition of hpS Tris-NaCl from three different placentas showing high amounts of glutamic/aspartic acid, and leucine (each around 10%).
Amino Acid Analysis of Human Placenta Substrate Tris-NaCl (Residues per 1,000 Residues) Compared with Other Extracellular Matrix Proteins from Literature
Amino acid quantification according to
hpS, human placenta substrate; SD, standard deviation.
A broad variety of natural polymers, already used for bioprinting in literature, were mixed with hpS Tris-NaCl to achieve a stable 3D solidification at 4°C or 37°C (Fig. 5).
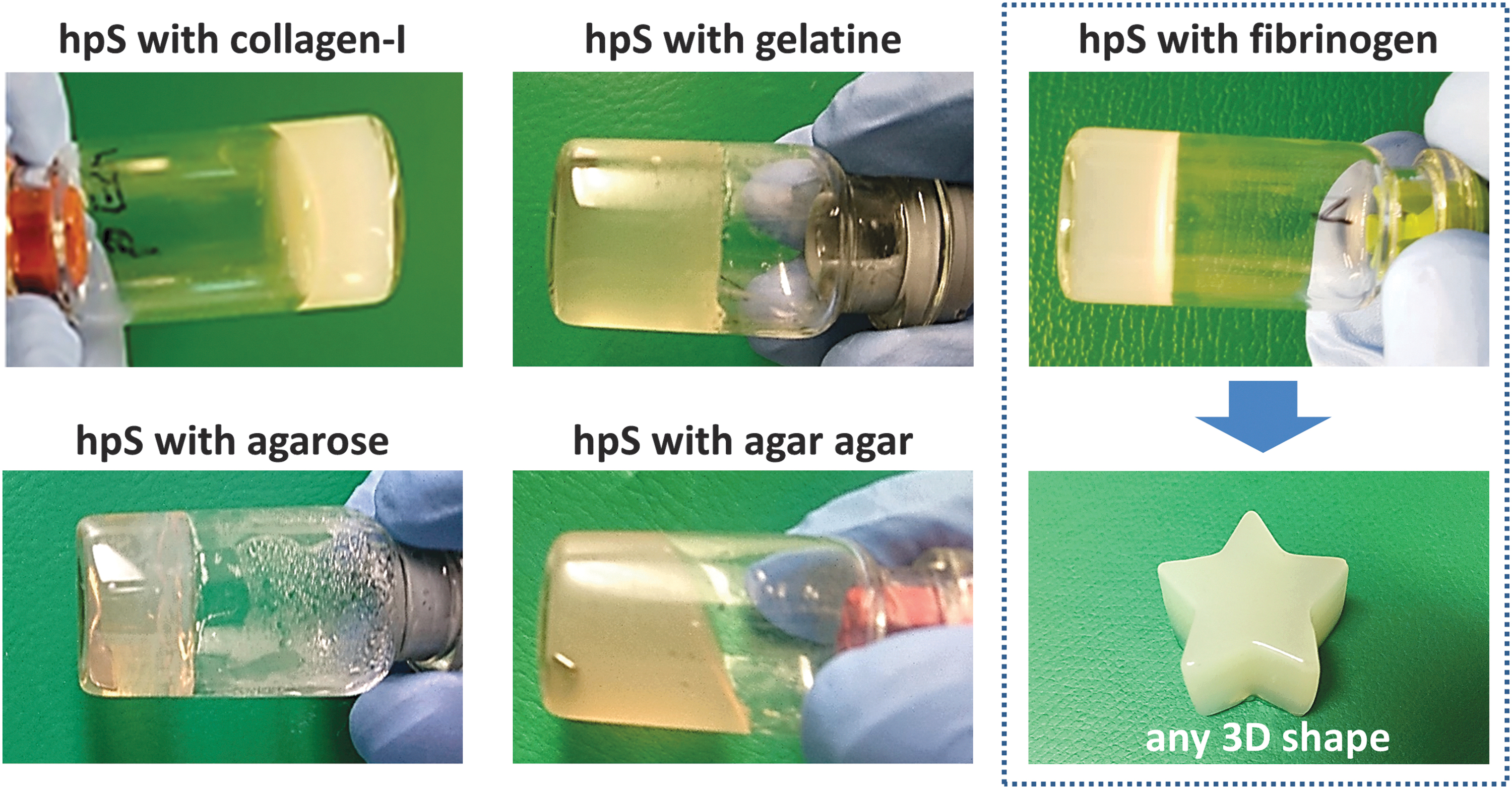
3D solidification of hpS. Various polymers were mixed with hpS to form stable 3D gels. As an example, hpS and fibrinogen were mixed without thrombin or aprotinin supplementation to gel at 37°C. 3D, three dimensional. Color images are available online.
2D biocompatibility of hpS
HUVEC seeding density
Different cell numbers (5,000–25,000 cells/well) were cultured for 2 days on hpS Tris-NaCl or hpS Tris-urea and the networks were analyzed (Fig. 6A–C). At seeding densities from 10,000 to 20,000 cells in 96 wells (30,000–60,000 cells/cm2), interconnected networks were formed in a cell number-dependent manner in the first 24 h of culture. Five thousand cells developed only partial cell networks and 25,000 cells yielded confluent nonpolarized cell layers that were not further analyzed. The network characteristics (total/mean tubule length, junctions) using 20,000 cells were significantly increased compared with all other cell seeding concentrations on both substrates, Tris-NaCl and Tris-urea (data not shown).
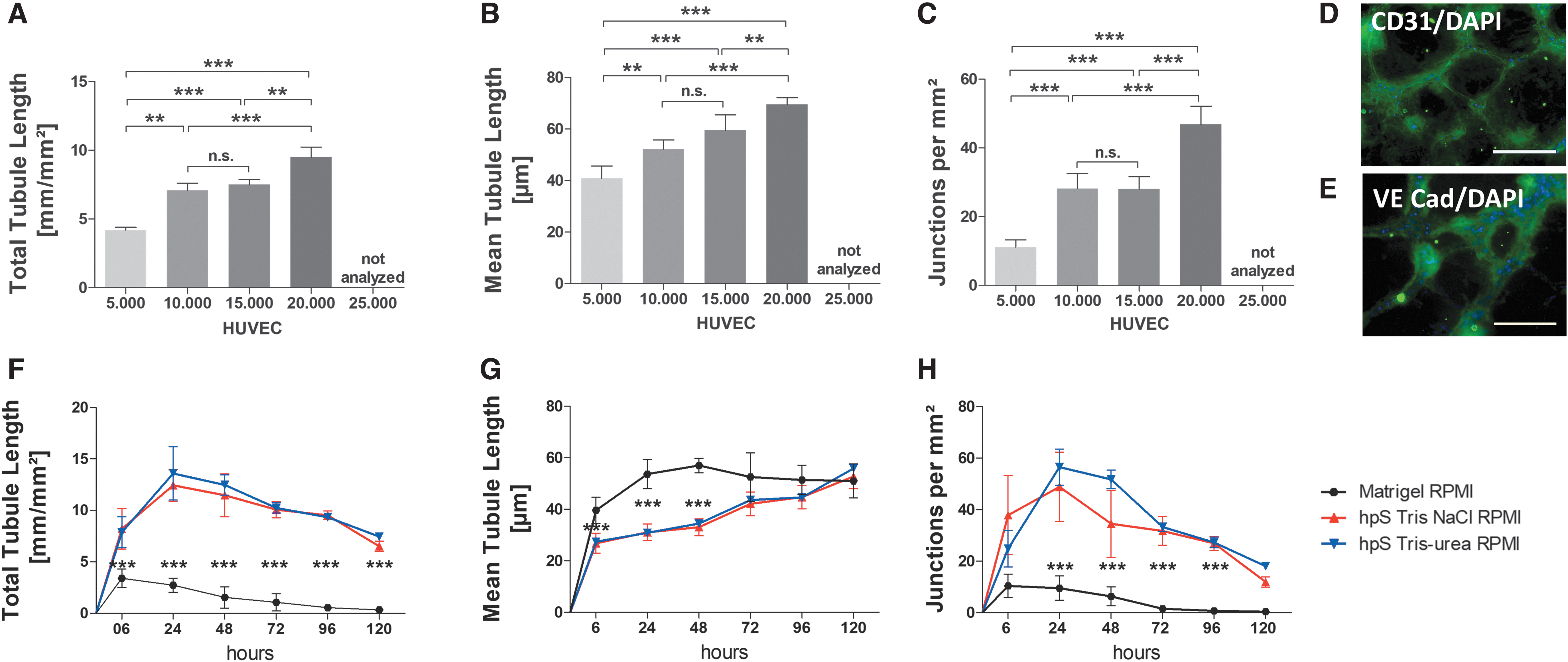
HUVEC seeding density on hpS-coated well plates in 2D.
Immunohistochemistry
CD31 and VECad, both markers for endothelial cells, were detected on HUVECs that assembled into an interconnected cell network (vasculogenesis) when seeded on hpS Tris-NaCl (Fig. 6D, E).
Comparison of substrates
Twenty thousand gfpHUVECs from the same donor were seeded on hpS Tris-NaCl, hpS Tris-urea, or Matrigel, and the cells were cultivated using minimally essential RPMI medium. The networks were analyzed after 6/24/48/72/96 and 120 h. On Matrigel, the network characteristics (total/mean tube length, number of junctions) were significantly lower when compared with both hpS. There were no significant differences in cell network characteristics between hpS Tris-NaCl and Tris-urea from the same donor using RPMI medium (Fig. 6F–H).
Single-placenta tissue comparison
Representative images of formed networks after 2 days are shown in Figure 7A. There was no significant difference observed in the network characteristics (total/mean tube length, number of junctions) between three different placentas, each weighing around 500 g (Fig. 7B), but the network characteristics were significantly increased when compared with Matrigel.
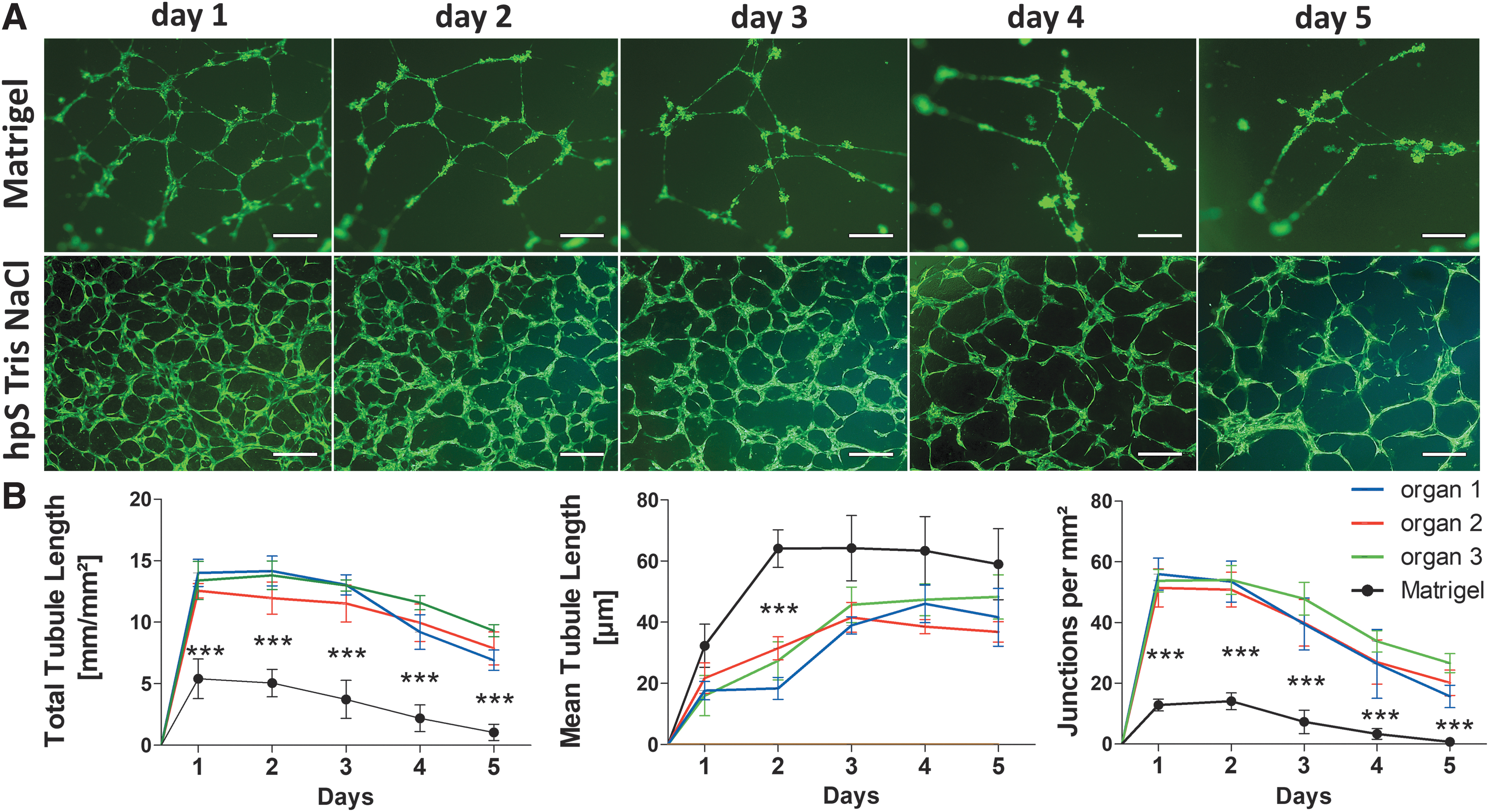
Single-placenta substrate compared in 2D.
gfpNIH3T3 fibroblast cultivation
Fibroblasts spontaneously formed networks when seeded on tumor-derived Matrigel, but not on hpS Tris-NaCl (Fig. 8A).
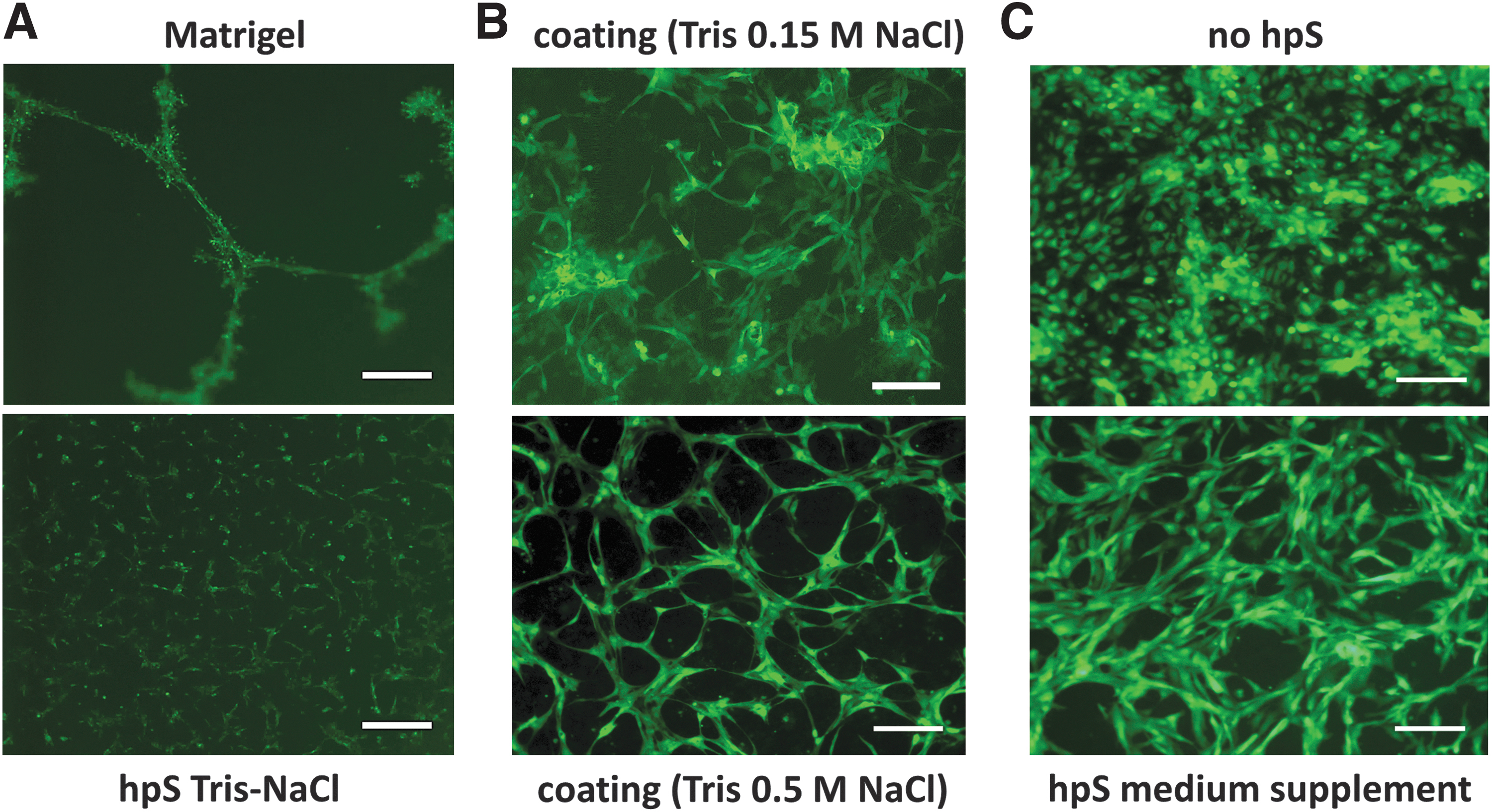
HUVEC/NIH3T3 fibroblast culture in 2D.
HUVEC culture supplementation
Substrates from human placenta extracted with a Tris 0.15 M NaCl buffer (physiologic) showed a different cell morphology and a lower in vitro performance when compared with hpS Tris 0.5 M NaCl (Fig. 8B). HUVEC polarization was also observed by applying hpS Tris-NaCl as a cell culture medium supplement, without hpS coatings (Fig. 8C).
3D biocompatibility of hpS
Fibrinogen hpS Tris-NaCl mixture
HUVECs seeded in a fibrinogen/hpS mixture formed a randomly orientated cell network with lumen, whereas HUVECs seeded in fibrin clots solidified with thrombin, no HUVEC network formation was observed (Fig. 9A). The microstructure of fibrinogen/hpS on SEM analysis showed a higher porosity in the hpS Tris-NaCl/fibrinogen clot when compared with the traditional fibrinogen/thrombin clot (Fig. 9B).
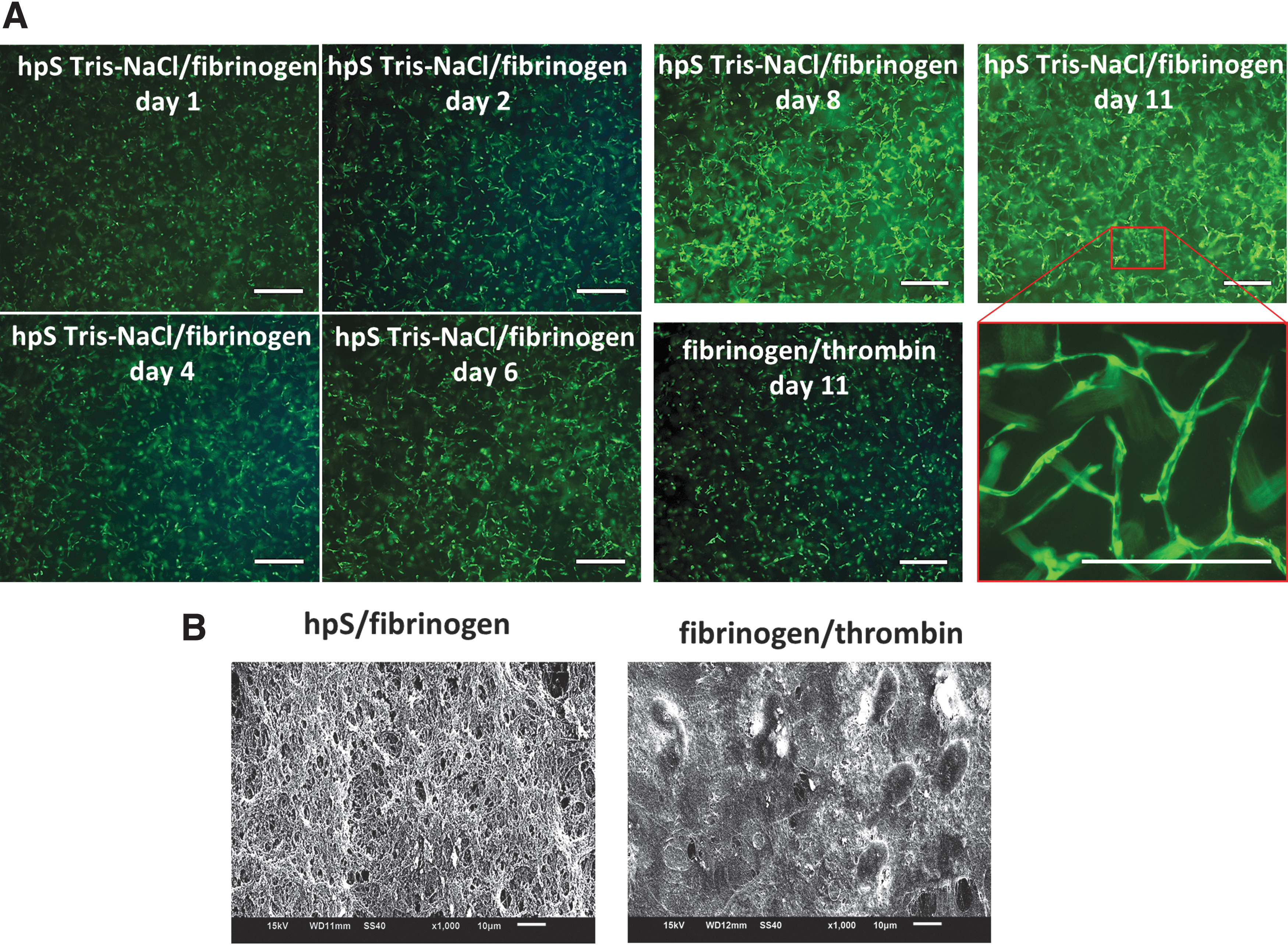
3D in vitro bioactivity of hpS.
Discussion
In the here presented study, we introduced the isolation of an effective method to isolate hpS (consisting of multiple proteins) from full-term human placenta, as a novel platform for a human/material-based technology for TERM.
Matrigel is originally extracted using a Tris 2 M urea buffer. 78 Various authors also used 2 M urea to isolate bioactive ECM from xenogenic tissues.8,79 Moore et al. used urea buffers ranging from 4 to 15 M to isolate a proangiogenic protein fraction from human placenta. 45 However, urea is an endogenous product of protein and amino acid catabolism primarily present in liver tissue, and the cancerogenic potential of urea has also still not been adequately assessed, due to the relatively few studies that have tested the toxicokinetics of exogenous urea in clinical studies to date. 80
Due to all these issues, Tris 0.5 M NaCl buffers were used in our experiments to isolate hpS, which are reported to preserve higher amounts of angiogenic cytokines compared with Tris-urea buffers if used for the preparation of tissue isolates.
This is in accordance with our findings that showed that in general higher amounts of tissue-inherent growth factors, cytokines and other angiogeneisis-related proteins and enzymes could be found in Tris-NaCl than in Tris-Urea hpS isolates. It has to be highlighted that this study did not include in vivo experiments and that still concerns related to in vivo use must be resolved beforehand as hpS represents a lysate of human placenta proteins including nucleic acids, which could cause immune reactions in vivo. Indeed, a fully decellularized ECM is regarded as a material with less than 50 ng/mL DNA dry tissue weight, DNA fragment sizes below 300 bp, and the absence of visible cellular particles, far away from the current DNA remnants in hpS. To solve this issue, Uriel et al. used an additional dispase treatment before starting the ECM extraction using a Tris 2 M urea buffer, to isolate proangiogenic ECM gels from dermis or fat tissue, with a final DNA content of 183.7 ± 10.2 ng/mL. 8
In a study by Choi et al., an enzymatic DNase/RNase treatment was used to lower DNA remnants from human placenta ECM-based extracts down to 34 ng/mg dry weight. 46 Nonionic detergents such as Triton X-100 are also often used to remove DNA from tissues,18,22,76–79 but Triton X-100 damages GAGs, which play an important role for cell attachment. Toxic ionic detergents such as SDS are also effective to remove cytoplasmic and nuclear cellular membranes from tissues.9,19,21,25,28,29 However, SDS tends to denature ECM proteins. 8 Its use requires intense washing steps to remove SDS from ECM, to prevent potential toxicological effects. 75
These strategies could be integrated in our presented isolation method to remove DNA in hpS for potential in vivo applications, however, it may also have an influence on its bioactivity, and therefore, more research is necessary before entering in vivo applications.
On average, 350 mL of liquid hpS (with a total protein concentration of 1.7 mg/mL in average) was extracted from one single placenta weighing around 500 g. Hence, our substrate could be used as a coating, injected into tissues, or soaked into any preexisting porous 3D materials for various cell culture applications. 45
The total protein concentration of hpS using a Tris 2 M urea buffer was significantly higher when compared with the Tris 0.5 M NaCl buffer, which might be the result of the higher ionic density. For instance, Moore et al. used a Tris 4 M urea buffer to yield a similar protein content to Matrigel (around 15–20 mg/mL).17,45 Hence, higher ionic densities yield higher amounts of ECM proteins. However, in the same way, they also seem to lower the amounts of residual bioactive growth factors (Fig. 3). 45 No significant differences of GAGs were detected in both hpS extracts when compared with native tissues.
Using SDS-PAGE, a heterogenic variety of separate protein bands ranging up to around 500 kDa were found in hpS Tris-NaCl, which may represent an accurate mimicry of the fully diversity of noncellular physiologic human tissue (ECM), whereas Matrigel from tumors is composed of less proteins (mainly laminin-111 and collagen-IV).
COL1 was only detectable in urea-enriched buffers (Matrigel, hpS Tris-urea), but not on hpS Tris-NaCl, as determined by Western blot analysis.
On angiogenesis arrays, higher amounts of various angiogenesis-related proteins were assessed using the isolation protocol based on a Tris 0.5 M NaCl buffer, when compared with the use of a Tris 2 M urea buffer, to extract hpS. Angiogenin, the most prevalent chemokine in hpS, was also the most prevalent chemokine using a Tris 4 M urea buffer in literature, but only relatively low levels of other angiogenic proteins were found. 45 Choi et al. used 0.5% SDS to extract ECM from human placenta and showed relatively high amounts of bFGF, TIMP-2, hepatocyte growth factor (HGF), or IGF binding proteins (IGFBP-1), but only relatively low levels of angiogenin were found. 46
In this regard, besides angiogenin, a heterogenous mixture of other angiogenic growth factors and chemokines led to the observed gfpHUVEC network formation on hpS. For instance, laminin-111 promotes angiogenesis in synergy with FGF-1 by gene regulation in endothelial cells. 81 Leptin, an endocrine hormone, stimulates angiogenesis in a synergistic effect with FGF. 82 Another prominent example is VEGF, known to play fundamental roles in the early phase of neovascularization (tip cell), whereas angiopoietin is associated with late-stage neovascularization (maturation of blood vessels).
Interestingly, hpS Tris-NaCl also contains thrombin, which upon mixing with fibrinogen can be used to form stable fully human 3D fibrin scaffolds (clots). In addition, hpS Tris-NaCl has also antimicrobial properties depending on the bacterial strain. The antibacterial effect was prominent in S. carnosus, whose growth was almost completely inhibited by hpS Tris-NaCl. Interestingly, other strains were not affected by hpS Tris-NaCl. However, the underlying mechanism has not been investigated so far.
The total amino acid analysis was used to identify the content of amino acids suitable for chemical crosslinking with other materials. The amino acid composition of hpS Tris-NaCl displayed relatively high contents of amino acids with modifiable side groups (around 20 mol% NH2/COOH residues), and we can therefore conclude that various chemical methods such as an anhydride strategy (e.g., norbornene anhydride), 83 NHS activation (e.g., allyl glycidyl), 84 or vinyl esters 85 can be used for functionalization of hpS and are currently studied.
Besides the characterization of the isolates we performed various experiments to show their usability in 2D as well as 3D cell culture applications. In our 2D in vitro assays, the cell network characteristics highly depended on the numbers of cells seeded, but not on different placentas (weighing each ∼500 g). In all experiments performed, a significantly higher network complexity was observed using hpS coatings (p < 0.001) when compared with Matrigel coatings. For instance, the mean tube length using hpS coatings reflects the physiological appearance (e.g., in a retina) in vivo, whereas the mean tube length is significantly longer when using Matrigel from tumor materials.
The interconnected cell networks on hpS remained for around 5 days in vitro, even when only using minimally essential RPMI medium, whereas the cell networks on Matrigel develop faster, but also degrade faster, as confirmed in literature. 45 There were no significant differences of the cell network characteristics observed on both hpS, although the total protein content in Tris-NaCl is around 25% lower than Tris-urea, and it contains a different protein composition.
The physiological relevance of Matrigel as a cell culture substrate is often called into question, as assays performed on Matrigel may result in false-positive and false-negative research results.68,86 For instance, in vitro, endothelial and also many nonendothelial cell types, such as NIH3T3-fibroblasts, melanoma, glioblastoma, breast cancer, or aortic smooth muscle cell lines, are already reported to form interconnected networks when seeded on Matrigel.16,45,87,88 Therefore, we performed an experiment using gfpNIH3T3 fibroblasts. While these cells did not form networks on hpS, they spontaneously formed networks on Matrigel within the first 24 h, which again confirms that Matrigel can also provoke false-positive or false-negative research results.
Using a physiological Tris 0.15 M NaCl buffer to precipitate hpS would substitute the remaining dialysis steps, however, the protein concentration and the final in vitro bioactivity were low when compared with our used Tris 0.5 M NaCl precipitation buffer. We could also show that hpS can also be used as a cell culture medium supplement. However, more studies using various cell types are currently under investigation to assess its full potential as an animal-free medium supplement.
After the 2D experiments, we translated our findings to 3D approaches since they are known to mimic the in vivo situation more accurately, when compared with 2D in vitro techniques. Indeed, many new technologies have been explored over the last years to pattern vascular cells in 3D hydrogels,89–92 and to guide vascular organization via chemical or mechanical signals.93–97 In addition, various publications have shown that channeled hydrogels improve the vascularization rate in 3D matrices.67,98 Hence, various fabrication techniques have already been utilized to create channel networks in hydrogels, including (1) removable structures, (2) 3D laser-assisted printing of photo-hydrogels, or (3) planar processing such as layer-by-layer UV radiation and polymerization of hydrogels.61,68,98
For our experiments, we mixed hpS with various natural proteins to form 3D hydrogels, to provide a useful material for many in vitro applications such as 3D cell culture, bioprinting, or perfused constructs.
For instance, in our 3D vasculogenesis studies, freeze-dried human fibrinogen, a clinically established product for decades, was mixed with hpS Tris-NaCl to induce a randomly oriented vasculogenic cell network in 3D after around 10 days of in vitro culture, whereas in traditional fibrin clots mixed with thrombin, no vasculogenic effects were observed within this time frame.
Conclusion
In summary, we could establish an effective method to isolate multiple proteins with angiogenesis-inductive properties from healthy human placenta tissue (hpS) with various potential applications for TERM. This material could be used as a novel platform for a human/material-based technology, for various 2D and 3D in vitro assays, as a medium supplementation, 3D bioprinting, and probably also for clinical applications.
Footnotes
Acknowledgments
The authors acknowledge the Red Cross Blood Transfusion Service of Upper Austria, Linz, for providing the placenta tissue. The authors thank Dr. Severin Mühleder for providing gfpHUVECs and antibodies to CD31/VECAD and Mag. med. vet. James Ferguson for reviewing the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by the Austrian Research Promotion Agency FFG (No. 849755) and, in part, by the City of Vienna Competence Team Project AgingTissue (MA23, No. 29-07).