
Abstract
The use of gelatin microspheres (GMs) as a cell carrier has been extensively researched. One of its limitations is that it dissolves rapidly in aqueous settings, precluding its use for long-term cell propagation. This circumstance necessitates the use of crosslinking agents to circumvent the constraint. Thus, this study examines two different methods of crosslinking and their effect on the microsphere's physicochemical and cartilage tissue regeneration capacity. Crosslinking was accomplished by physical (dehydrothermal [DHT]) and natural (genipin) crosslinking of the three-dimensional (3D) GM. We begin by comparing the microstructures of the scaffolds and their long-term resistance to degradation under physiological conditions (in an isotonic solution, at 37°C, pH = 7.4). Infrared spectroscopy indicated that the gelatin structure was preserved after the crosslinking treatments. The crosslinked GM demonstrated good cell adhesion, viability, proliferation, and widespread 3D scaffold colonization when seeded with human bone marrow mesenchymal stem cells. In addition, the crosslinked microspheres enhanced chondrogenesis, as demonstrated by the data. It was discovered that crosslinked GM increased the expression of cartilage-related genes and the biosynthesis of a glycosaminoglycan-positive matrix as compared with non-crosslinked GM. In comparison, DHT-crosslinked results were significantly enhanced. To summarize, DHT treatment was found to be a superior approach for crosslinking the GM to promote better cartilage tissue regeneration.
Impact statement
Gelatin microspheres (GMs) have been thoroughly investigated as a cell delivery system. However, gelatin disperses rapidly in aqueous environments, making the use of polymers in the development of long-term delivery systems problematic. In the formation of insoluble networks in microspheres, this unfavorable factor necessitates the use of a crosslinking agent. To address this issue, GMs were crosslinked using a naturally and physically occurring crosslinking agent (dehydrothermal [DHT] and genipin) as a biodegradable cell delivery tool for intra-articular administration. DHT was identified as the most favourable crosslinker for GM for cartilage regeneration among two separate crosslinking agents.
Introduction
Three-dimensional (3D) porous scaffolding can be implemented to promote bone and cartilage regeneration.1–4 The scaffolds must have a bioactive surface, adequate microarchitecture, and mechanical stability for cells to colonize and form new tissue. 5 Collagen is the most promising biomaterial for tissue regeneration since it constitutes the majority of the extracellular matrix (ECM) in the body. Previous research has demonstrated the efficacy of using equine and ovine tendon type I collagen as a self-assembling 3D matrix to stimulate osteochondral tissue regeneration.6,7
Gelatin, a form of hydrolyzed collagen, is a good alternative due to its similarity to collagen in terms of its low cost, nonimmunogenicity, high biocompatibility, and good biodegradability3,8–11 and the added advantage of its gelation property. However, compared with other synthetic gelable polymers, there are certain drawbacks to using gelatin, such as limited elasticity, heat instability, and high solubility in water. 12 To overcome this, gelatin is frequently stabilized by inducing chemical interactions among their exposed functional groups.12,13
Chemical cross-linkers such as carbodiimide hydrochloride, 1-ethyl-3-(3-dimethylamino propyl), glutaraldehyde, and 1,4-butanediol diglycidyl ether can provide the reinforcement required. Physical crosslinkers such as dehydrothermal (DHT), ultraviolet, and gamma irradiation can also be used.10,14 Genipin, a chemical crosslinker, is an iridoid glucoside that may cross-link proteins by intra- and inter-molecular interaction, and is commonly used in tissue engineering applications to create tissue-specific biomaterials.15–17
Microspheres are known for their role as a carrier in drug delivery systems and as a 3D scaffold for in vitro cellular expansion.18–20 Recently, several reports demonstrated their safety and efficacy in delivering cells for cartilage regeneration in vivo.21,22 The microsphere is the contact point for cell attachment and growth, ensuring cell viability while providing a foundation for native cells to invade and regenerate.3,10 Our recent study reported that gelatin microspheres (GMs) increased the chondrogenic differentiation of mesenchymal stem cells (MSCs).3,10,11 As microspheres are minuscule, the delivery does not require open surgery as they can be injected percutaneously into the intra-articular space. 23 However, gelatin dissolves rapidly in aqueous environments limiting the polymer's usage for long-term delivery systems creating the need to exploit the crosslinking agent to overcome this limitation.
This study scrutinizes the physicochemical and biological features of GMs as cell microcarriers for the regeneration of cartilage tissues. 3D gelatin matrices were stabilized using two crosslinking methods (DHT and genipin) to create chemically and structurally reinforced biocompatible scaffolds. Water uptake, biodegradability, and thermal behavior of the 3D microspheres were compared between the different crosslinking treatments. Chondrogenic indicators were examined utilizing bone-marrow-induced chondrocytes in in vitro biological assays to determine cell survival, proliferation, morphology, and gene expression.
Materials and Methods
In this study, 3D gelatin matrices were crosslinked using DHT and genipin to create reinforced and chemically stabilized biocompatible scaffolds for cartilage tissue engineering applications. To determine the effect of the various crosslinking treatments on the water uptake, biodegradability, biocompatibility of the 3D scaffold, a comparative evaluation was performed. In addition, human bone marrow stromal (stem) cells were used in in vitro biological tests to assess cell viability, proliferation, and morphology, as well as gene expression for chondrogenic markers (Fig. 1).

Schematic diagram of the experimental model of gelatin microsphere production, crosslinking, and characterization process.
Experiment
Preparation of GMs
GMs were prepared using the water-in-oil emulsion method.10,11 Twenty milliliters of 10 wt% gelatin solution (Nitta Gelatin Inc., Osaka, Japan) were heated to 40°C and added dropwise into 600 mL of olive oil (Wako Ltd., Osaka, Japan). The mixture was stirred at 400 rpm for 10 min to formulate a water-in-oil emulsion. The produced GMs were washed 3 × with cold acetone combined with centrifugation (5000 rpm, 4°C, 5 min). The size of GMs was fractionated using sieves with apertures of 20, 32, and 53 μm (Endecotts Ltd., London, United Kingdom) and air-dried at 4°C.
DHT-crosslinked GM (GM-DHT): the non-crosslinked and dried GM (200 mg) was treated in a vacuum oven at 140°C and 0.1 torr for 72 h.10,11,24
Genipin-crosslinked GM (GM-GEN): the gelatin solution was added with 0.5wt% of genipin (Wako, Germany) and mixed by stirring for 5 min. The resultant mixture was transferred into a polystyrene Petri plate, and the crosslinking reaction was carried out at 25 ± 2°C for 72 h. 25
Crosslinking of GM
A ninhydrin assay was performed to assess the percentage of uncrosslinked amino groups, which is used to calculate the degree of crosslinking in the manufactured GM (Sigma Aldrich, Saint Louis, MO). After 24 h of lyophilization, the test samples were weighed and heated for 2 min at 100°C in ninhydrin solution. The number of free amino groups in the test sample was evaluated using a spectrophotometer at an absorbance of 570 nm (BioTek, PowerWave XS, Highland Park, IL). In addition, various concentrations of glycine (1.0, 2.0, 3.0, 4.0, and 5.0 mg/mL) were used to generate a standard glycine concentration versus absorbance curve.
The number of free amino groups can be determined using the standard curve. The crosslinked GMs were then rinsed with aqueous ethanol solution (99.5% ethanol by volume) to remove residual genipin for 4 h. Subsequently, the rinsed microspheres were vacuum-dried for 24 h to evaporate ethanol. The crosslinked microspheres were sprinkled onto a double-sided adhesive tape fixed to an aluminum stage. The fixed microspheres were spattered with gold film (Sputter Coater Q150TES, Quorum, Italy). Examination of microspheres was performed with a scanning electron microscope (SEM; FEI, Quanta 200, Universiti Kebangsaan Malaysia [UKM]).
Dynamic swelling of GM
Dynamic swelling of DHT- and genipin-crosslinked GM was carried out according to the method described by Robert et al.
26
On the observation plates equipped with liquid containers, dried test microspheres were inserted and seen using an optical microscope (IX70, Olympus Optical Co., Ltd., Tokyo, Japan). Their initial diameter, d0, was recorded, and deionized water was added to the containers. At room temperature, the increase in the swelling diameter, dt, due to water transport in test microspheres was measured as a function of time, until the microspheres reached equilibrium with a diameter of d∞. For each study group, six microspheres with diameters between 50 and 100 μm were evaluated. The mechanism of water transport can be deduced by examining water uptake curves as a function of time using the following Equation (1):
26
(dt – d0)/d0 = fractional amount of water uptake
t = water transport time
k = diffusional kinetic constant
n = diffusional exponent.
When analyzing transport data for (dt – d0)/d0 <0.6, the value of the diffusional exponent (n) is a good indication of the water transport mechanism.26,27
Enzymatic degradation of GM
Enzymatic degradation of the DHT- and genipin-crosslinked GM was performed using 0.0006% (w/v) type I collagenase (Worthington, Lakewood, NJ). Degradation of test microspheres without collagenase was used as a control. Test microspheres were well immersed in the collagenase solution (pH 7.5) and incubated at 37°C for 24 h. Degradation of test microspheres was discontinued at the desired duration by adding a 10 mM ethylenediaminetetraacetic acid (EDTA; Sigma E9884) solution followed by rapid cooling to 4°C. The morphology of test microspheres after enzymatic degradation was grossly examined using a SEM as described above. The GM was then freeze-dried to estimate its dry weight. The dry weight of the GM was determined every hour up to 14 h or until total deterioration.
Physicochemical analysis of GM
Chemical analysis through Fourier-transform infrared spectroscopy
The infrared spectra of the scaffolds were obtained using Nicolet 380 FT-IR spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Two milligrams of the scaffold were mixed with 100 mg of anhydrous KBr and pressed at 8000 psi into 7 mm diameter disks. The spectra were collected in the wavelength ranging from 400 to 4000 cm−1 with 2 cm−1 of resolution.
Cell isolation and culture
Ethics approval for this study was granted by Universiti Kebangsaan Malaysia Research Ethics Committee with the approval code UKM PPI/111/8/JEP-2018-458 on October 11, 2018. Bone marrow samples were collected from six patients (n = 6) undergoing a total knee replacement procedure with their prior written consents. Five to ten milliliters of bone marrow aspirates were obtained from the knee joints of the patients (54–69 years). The collected bone marrow mesenchymal stem cells (BMSCs) were isolated and cultured in a tissue culture plate (TCP) according to the method previously described. In brief, the samples were processed within 6–12 h after collection. BMSCs were isolated using the Ficoll-Paque method.
The BMSCs were then cultured in FD (F12: Dulbecco's modified Eagle's medium [1:1] supplemented with 10% fetal bovine serum [FBS; Biowest, Riverside, MO], 1% antibiotic-antimycotic [Gibco, Grand Island, NY], 1% glutamax [Gibco], and 1% vitamin C [Sigma-Aldrich]). The cells were incubated at 37°C in a humidified atmosphere with 5% carbon dioxide (CO2). The culture medium was changed every 2–3 days until 70%–80% confluency. Cells were trypsinized using 0.05% Trypsin–EDTA (Gibco) and subcultured until passage 3. Passage 3 cells were centrifuged, and resuspended in 5 mL of the medium before cell count and used for subsequent experiments.
To evaluate the response of the GMs, BMSCs monolayer and cell micromass (BMSCs-GM-DHT and BMSCs-GM-GEN) in 3D suspension were cultured for 14 days in chondrogenic media, and analyzed for chondrocyte markers through gene and protein expression, growth kinetics, and proteoglycan analysis. Cells used for immunophenotyping were cultured separately using FD medium until day 14. All cultivation was done in the hypoxic incubator (5% oxygen).
Construction of cell micromass
GMs were sterilized with 70% ethanol, followed by complete washing with sterilized phosphate buffer saline (PBS; Sigma-Aldrich). Before transferring the GM onto the TCP, the plates have to be coated with polyvinyl alcohol (PVA; Sigma-Aldrich, polymerization degree of 1800, and 88 mole % saponification) to prevent cell attachment onto the plate. In brief, 1% PVA in PBS was added into each of 12- and 24-well plates (1 mL/well) and incubated at 37°C for 15 min. Then, the solution was removed by aspiration, and the well was washed with PBS (1 mL/well) twice.
For cell differentiation experiments, GMs were transferred into 12-well plates at 10 mg/well, and 5 × 104 BMSCs at passage 3 were seeded onto the microspheres per well (i.e., 5 × 103 cells/mg of microspheres), and cultured in a chondrogenic medium in a hypoxic incubator at 37°C and 5% CO2. The chondrogenic medium contains FD medium (Gibco) supplemented with serum, 1% Insulin Transferring Selenium (Gibco), 0.2 mM ascorbic acid-2 phosphate (Sigma), 40 ng/mL L-proline (Sigma), 100 nM dexamethasone (Invitrogen, Inc., USA), 10 ng/mL transforming growth factor-beta 3 (Invitrogen Inc.), and 50 ng/mL insulin-like growth factor 1 (Invitrogen Inc.).28,29 The medium was changed twice a week.
On day 14, the cell micromass were harvested and processed for toluidine blue and immunohistochemistry staining for type II collagen, and lysed for gene expression analyses. For cell proliferation experiments, GMs were transferred to 24-well plates at 2 mg/well, and 1 × 104 cells/well were subsequently seeded onto the microsphere. The same number of cells was cultured on non-PVA-coated TCP as control, which was later compared with the microsphere group in both experiments.
Cell viability and proliferation assay
Cell viability was assessed by live/dead (Invitrogen) assay. On days 1, 7, and 14, cell micromass were washed in PBS (pH 7.4) for 5 min, and stained with 2 μM of calcein and 4 μM of EthD-1 for 15 min at 37°C. The scaffolds were washed in PBS (pH 7.4) for 5 min and viewed under an inverted Ti-E fluorescence microscope (Nikon, Japan). Presto Blue (Thermo Fisher Scientific) was used to study the proliferation of the cell micromass. All the groups were cultured in FD for 14 days in replicates, and the data were collected at different time points for analysis (days 1, 7, and 14). All the respective culture and blank plates were rinsed with PBS, and then 180 μL of FBS-free medium was added into each plate followed by 20 μL of Presto Blue reagent on top of the FBS-free medium.
Later, the plates were incubated for 2 h at 37°C and 5% CO2. After 2 h of incubation, 100 μL of the supernatant was transferred into a 96-well plate, and, subsequently, absorbance was measured using a microplate reader at 570 nm against a reference wavelength of 600 nm. The experiment was performed in triplicates. Hoechst staining at the manufacturer-recommended concentration of 1 g/mL was utilized to visualize living cells. The cells' medium was withdrawn and replaced with a medium containing a dye. The cells were cultured at room temperature or 37°C for 5–15 min, and then observed and analyzed using a fluorescence microscope.
Cell morphology
After 1, 7, and 14 days, the cell-seeded scaffolds were washed with 0.1 M sodium cacodylate buffer (pH 7.4) and fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 h at 4°C. The scaffolds were rewashed in the same buffer, freeze-dried, and mounted onto the aluminum stubs using black carbon tapes and sputter-coated with gold particles as mentioned above. The morphology of the cell micromass was examined using high-resolution SEM.
Immunophenotype analysis
The immunophenotype analysis was used to investigate the characteristics of BMSCs cultured on GM-GEN and GM-DHT (BMSCs-GM-GEN and BMSCs-GM-DHT). Both groups were digested with 0.05% trypsin EDTA to obtain single cell suspension. The cells were then washed with 0.2% bovine serum albumin (BSA) in PBS, and stained with mouse antihuman CD73, CD90, CD105, CD14, CD34, and CD45 (BD Pharmingen, San Jose, CA). In brief, one million cells were suspended in 100 μL of 0.2% BSA in PBS and stained with individual antibodies at the manufacturer-recommended concentration for 30 min. The cells were washed twice in PBS, suspended in 0.2% BSA/PBS, and analyzed for stem cell surface marker expression using FACS Calibur cytometer (BD Biosciences, San Jose, CA) and Cell Quest Pro software. Ten thousand gated events were recorded. 3
Histology and immunofluorescence staining
The samples were washed with Dulbecco's phosphate buffered saline and fixed with 4% paraformaldehyde overnight, then stained with 0.04% toluidine blue to demonstrate the presence of proteoglycan in the samples. For immunofluorescence staining, the samples were permeabilized for 20 min with 0.5% Triton X-100 solution (Sigma-Aldrich), and then blocked with 10% goat serum for 1 h at 37°C. The cells were incubated with mouse antihuman collagen II (DSHB Cat:II-II6B3, RRID:Ab 528165) for chondrocytes overnight at 4°C. The following day, the cells were incubated with Alexa Fluor 594 goat antimouse antibodies (Invitrogen) for 1 h at 37°C and counterstained with 2-(4-amidinophenyl)-6-indolecarbamidine (Dako, Glostrup, Denmark) for 15 min. The cells were observed and evaluated with z-stack acquisition using a Nikon Eclipse Ti fluorescence microscope (Nikon).
Glycosaminoglycan assay
At the end of days 1, 7, and 14, the cell micromass were treated in lysis buffer (5 mg/mL papain in 50 mM sodium phosphate buffer (pH 6.4) containing 5 mM EDTA and 5 mM cysteine) for 3 h at 65°C. The digestive solution was collected, centrifuged, and analyzed for sulfated glycosaminoglycan (sGAG) using dimethyl methylene blue (DMMB) dye and read using a spectrophotometer. Twenty microliters of sample and 200 μL of DMMB were pipetted into the microplate reader. Samples were analyzed by measuring the absorbance at 525 nm. 30
Quantitative real-time polymerase chain reaction
The total RNA was isolated from BMSCs and cell micromass after 14 days using the Qiagen RNeasy mini kit. 3 The concentration of total RNA was determined using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized (Thermo Scientific) using the Maxima first-strand cDNA synthesis kit. Quantitative real-time polymerase chain reaction (qPCR) was performed on the cDNA using the SYBR FAST Biorad qPCR master mix (Bio-rad, Hercules, CA) with primers listed in Table 1. The delta–delta Cycle Threshold (CT) value method calculated the relative fold change in gene expression. The average CT value was determined for each sample in triplicate. After normalizing the CT value to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), the delta CT value was computed. The relative fold change was calculated as follows:
Primer Sequences Used for Real-Time Polymerase Chain Reaction Experiments
where ΔCT = (CT target gene − CT GAPDH)
ΔΔCt = ΔCt (sample) − ΔCt (control).
Statistical analysis
Results were plotted on a graph and expressed as mean ± standard error of the mean. Two-way analysis of variance was used to analyze cell proliferation, gene expression, and glycosaminoglycan content, followed by Tukey's multiple comparisons test. Statistical analysis was performed by Graph Pad Prism software (v8.0), with a statistical significance of p ≤ 0.05.
Results
Preparation of GMs
The GMs produced were smooth, spherical, and consistent in size (Fig. 2A–D). When submerged in water, they swelled and became translucent. The addition of DHT or genipin to the GMs had no noticeable influence on their shape (Fig. 2A, B). DHT-crosslinked GMs appeared yellowish white, whereas genipin-crosslinked GMs were dark blue in color.

Optical microscope images of GMs in wet conditions
The Fourier-transform infrared spectroscopy (FTIR) spectra of the produced scaffolds exhibit three distinct gelatin peaks (Fig. 2E): Amide I at 1690 cm−1 due to C-O stretching vibrations, Amide II at 1560 cm−1 due to N-H deformation, and Amide III at 1250 cm−1 due to C-N stretching vibrations. 31 No apparent vibrational alterations were seen in the spectral peaks of GM-DHT, and the spectrum appeared to be identical to that of the parent molecule.
The GM-GEN revealed a spectrum peak at 1650 cm−1, corresponding to the C = N stretching of gelatin amine groups, leading to creating a secondary amide through carboxymethyl group interaction with genipin. Another vibrational change at 1100 cm−1 reveals the stretching vibration of the dihydropyrane ring in genipin (heterocyclic ring opening), which is necessary for successful gelatin crosslinking. 32 The FTIR data indicate that the inclusion of crosslinkers significantly altered the chemical characteristics of the scaffolds without compromising their biocompatibility.
Figure 2F shows the degree of crosslinking for DHT- and genipin-crosslinked GMs (GM-DHT [GD] and GM-GEN [GG]) obtained at various crosslinking durations. After 72 h, the highest degree of crosslinking for GM-DHT was 60%; however for GM-GEN, it takes 2 h to attain 60% crosslinking. Thus, for the remainder of the investigation, the degrees of crosslinking for both GM-DHT and GM-GEN were set to 60%.
Dynamic swelling of test microspheres in deionized water was performed at room temperature. The fluctuation in the normalized particle diameter over time for GM-DHT and GM-GEN is depicted in Figure 2G. The normalized particle sizes of both test microspheres increased significantly in the first 5 min (p < 0.05) and then stabilized after 5 min in deionized water. GM-GEN had a larger equilibrium diameter (d) than GM-DHT. Equation (1) can investigate the water transport process in test microspheres. Figure 2G shows that the diffusional exponent (n) in Equation (1) was 0.47 and 0.57 for GM-DHT and GM-GEN, respectively (r 2 = 0.99).
The stability of fabricated GM
When subjected to enzymatic hydrolysis, the GMs degraded completely in less than an hour without crosslinking. GMs crosslinked with 0.5% genipin, and DHT resisted enzymatic hydrolysis for 6 and 7 h, respectively, before degradation can be observed (Fig. 3B). However, GMs crosslinked with 0.5% genipin and DHT withstood hydrolysis for 10 and 14 h without enzymatic hydrolysis.

Representative SEM micrographs of the GM-DHT and GM-GEN at 4 h after degradation with or without collagenase
Cell viability and proliferation assay
With increasing culture time, cell density increased, particularly on days 7 and 14, and the cells appeared to be evenly distributed across both scaffold surfaces. In addition, Hoechst 33242 staining in both scaffolds revealed round cell nuclei seen in dark blue on day 1 of culture (Fig. 4A), and cell colonization of the material increased with time (Fig. 4B). The proliferation of BMSCs on GM-GEN and GM-DHT scaffolds in 3D suspension was detected after 1, 7, and 14 days. In all designated time points, the GM-DHT group had significantly more cell proliferation compared with the GM-GEN group (p < 0.05, p < 0.001; Fig. 4C).
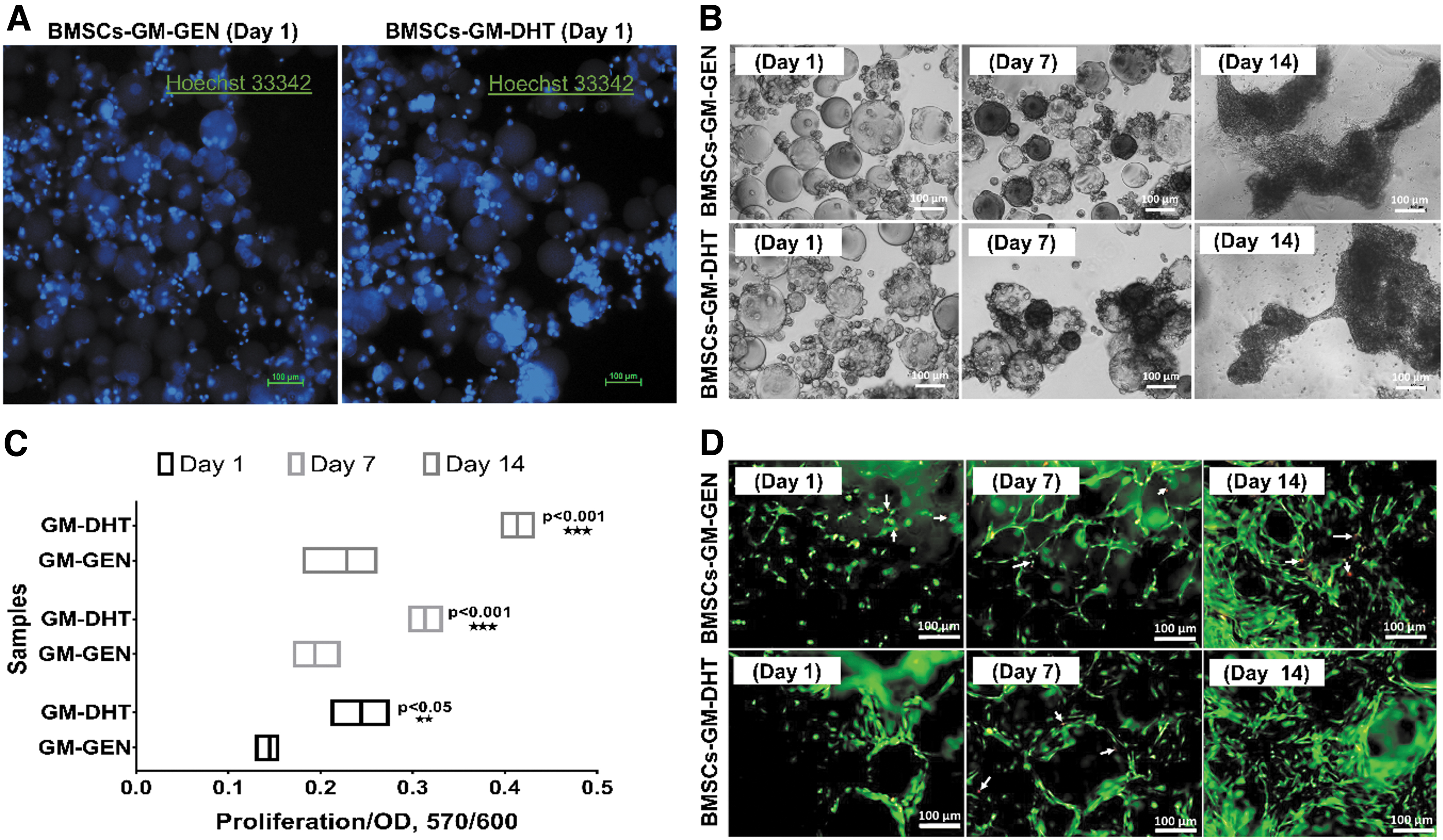
Cell viability and proliferation assay. Hoechst 33242 staining in both scaffolds revealed appropriate round cell nuclei morphology in dark blue on day 1 of culture
In Figure 4D, the live/dead assay of GM-GEN and GM-DHT scaffolds exhibits excellent cell viability at each time point, indicating good cytocompatibility. The live/dead and Hoechst 33342 staining images showed a steady increase in cellularity with increasing culture time, demonstrating that the scaffolds are not cytotoxic. No change in cell survival was observed between the scaffolds, and crosslinkers did not affect BMSC expansion or scaffold colonization.
Immunophenotype analysis and gene expression analysis
There was no significant difference in morphology and phenotype of BMSCs-GM-GEN and BMSCs-GM-DHT (Fig. 5A). The immunophenotype analysis was used to investigate the characteristics of BMSCs cultured on GM-GEN and GM-DHT (BMSCs-GM-GEN and BMSCs-GM-DHT). All BMSCs expressed MSCs surface markers CD73, CD90, and CD105 and lacked hematopoietic cell markers CD14, CD45, and CD34 (Fig. 5B). The relative expression of anabolic (sox 9, type II collagen, COMP, and ACAN) and catabolic genes (MMP13 and ADAMTS5) was compared between BMSCs-GM-GEN and BMSCs-GM-DHT after 14 days of culture (Fig. 5C) with BMSCs monolayer as control.
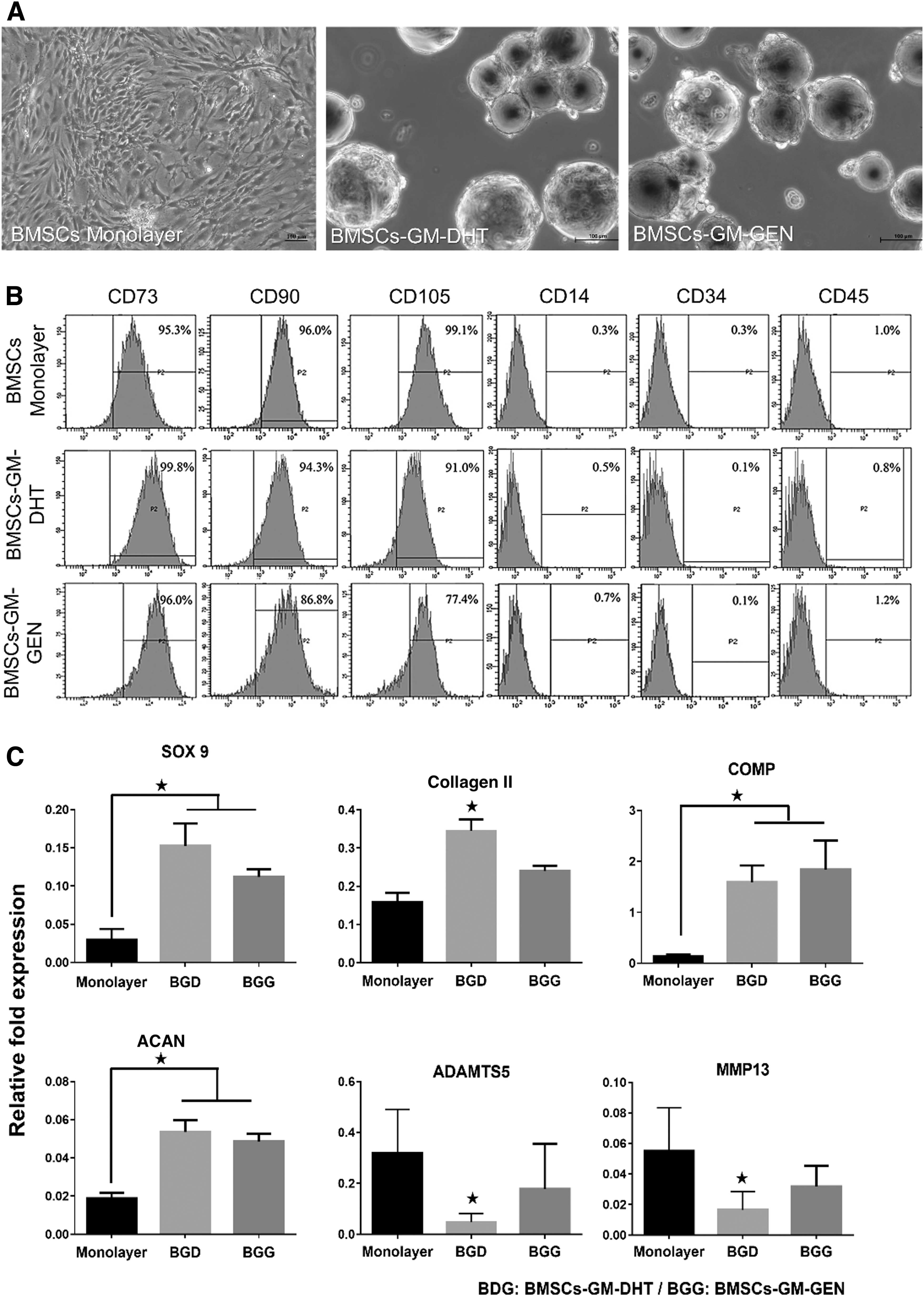
lmmunophenotype of cultured BMSCs in three different groups (monolayer [control] and cell micromass [BMSCs-GM-DHT and BMSCs-GM-GEN]) analyzed by flow cytometry. Cell morphology in three different groups at day 14 of culture
Both BMSCs-GM-DHT and BMSCs-GM-GEN showed significantly higher relative fold expression than monolayer culture for all anabolic genes. However, the expression of anabolic genes, except for COMP, was still higher in BMSCs-GM-GEN than in BMSCs-GM-DHT, but no significant difference was observed. Meanwhile, the expression of catabolic genes was found significantly lower only in BMSCs-GM-DHT as compared with monolayer (*p < 0.05) (Table 2).
Relative Fold Expression of the Anabolic and Catabolic Gene in BMSCs-GM-DHT and BMSCs-GM-GEN Compared with Monolayer Culture
p < 0.05.
BMSCs, bone marrow mesenchymal stem cells; DHT, dehydrothermal; GEN, genipin; GMs, gelatin microspheres.
ECM deposition
Microscopic investigations revealed that cells were dispersed uniformly on both BMSCs-GM-DHT and BMSCs-GM-GEN. ECM synthesis began on day 1 of culture with increased aggregation on days 7 and 14. SEM images showed that BMSCs attached and extended in response to ECM synthesis (Fig. 6A). After 14 days, the BMSCs deposited an abundant amount of ECM proteins on the surface of the microspheres, and displayed cell–cell and cell–microspheres interaction (Fig. 6B). The histological study suggested that GMs promote matrix formation and type II collagen expression (Fig. 6C). The amount of sGAG generated by the cells increased significantly on days 7 and 14 for BMSCs-GM-DHT compared with BMSCs-GM-GEN (Fig. 6D). Thus, the increased glycosaminoglycans produced by the cells reflect the chondrocytes' extensive deposition of cartilage ECM components.
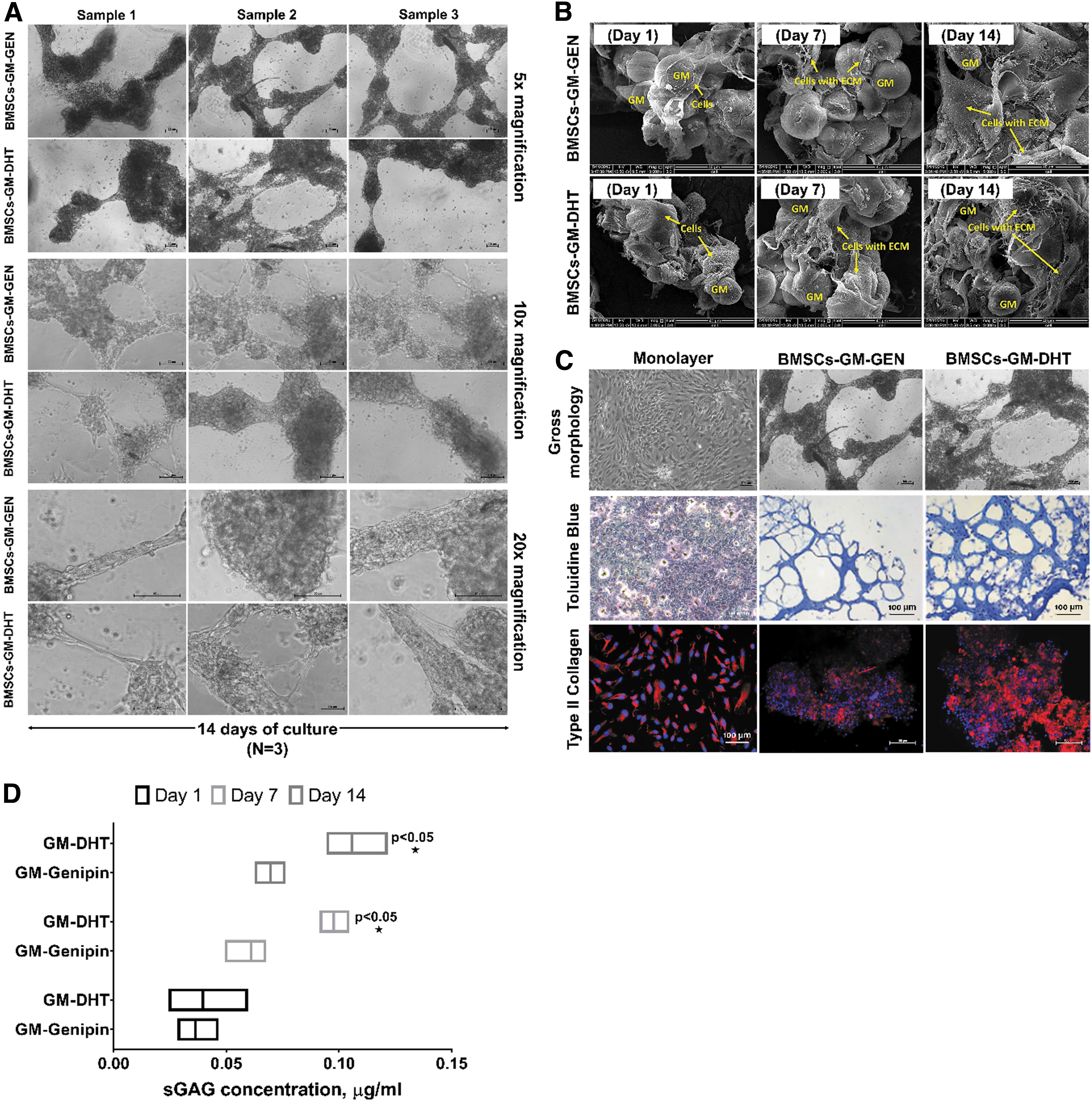
The extracellular matrix deposition on gelatin microspheres was monitored from days 1 to 14. On day 14, BMSCs aggregated and produced ECM homogenously on GM-DHT and GM-GEN. The figures were magnified 5 × , 10 × , and 20 ×
Discussion
This study compares the performance of two different crosslinking agents, that is, DHT and genipin, on GM. The amount of crosslinkers used is based on the preliminary screening done earlier.11,24,25 To select the best crosslinking proportion, different parameters of genipin and DHT were prepared. The optimum amount of crosslinker (genipin [0.5 wt%] and duration of DHT [48 h]) was chosen based on the minimum concentration of genipin required to obtain a 3D scaffold with good physical stability.
The DHT treatment is a physical process that includes repeatedly applying high temperatures under a vacuum (generally >100°C and <100 mTorr) (typically a few days). Under these conditions, water is removed from the gelatin as a result of thermal dehydration. This allows the amino and carboxyl groups of gelatin molecules to come close to one another to develop chemical bonds through esterification or amide link formation.33–35
The dark bluish color of the genipin-crosslinked microspheres was produced when genipin reacted with amino acid residues in gelatin molecules. It was hypothesized that the third carbon of genipin can act on an amino-group-containing substance, such as gelatin, to produce a genipin-amino-group monomer through a nucleophilic reaction. This is followed by the opening of the genipin ring, resulting in the formation of an aldehyde group, which can react with the attached secondary amino group. Dimerization then occurs, possibly as a result of radical reaction, and produces heterocyclic compounds with intramolecular and intermolecular crosslinking.35,36 Thus, both DHT and genipin crosslink the amine group within the gelatin.
Crosslinking enhances the structure of a polymer and protects it against breakdown. 25 The degree of crosslinking determines the stability of synthesized GMs. The degree of crosslinking impacts the stability of GMs that have been synthesized. Therefore, detection of the free amine group using the ninhydrin assay shows the amount of polymeric network crosslinked in a bioscaffold. 25 This parameter, known as the degree of crosslinking, is essential as the higher the crosslink, the higher the scaffold's structural integrity. An increasing trend for the degree of crosslinking was observed following an increase in incubation time of crosslinking agents.
In the study, the maximum degree of crosslinking for GM-DHT after 72 h was 60%, whereas for GM-GEN it takes 2 h to achieve 60% crosslinking. Thus, during the remainder of the investigation, the degrees of crosslinking for both GM-DHT and GM-GEN were set to 60%.
Degradation test was performed to assess the scaffolds' stability and mass loss over time under physiological conditions. Under microscopic observation, the scaffolds changed their shapes or had an altered structural architecture during the degradation test. Non-crosslinked scaffolds dissolved in less than an hour when subjected to enzymatic hydrolysis at 37°C. Other researchers have reported similar results.25,37
A slow degradation rate can be attributed to the material's high degree of crosslinking. However, despite the similar degree of crosslinking for GM-DHT and GM-GEN tested in this study GM-DHT had a lower dissolution rate compared with GM-GEN. Thus, besides the degree of crosslinking, the type and strength of covalent bonding initiated by treatment with genipin and DHT are likely different. It is also important to note that the time required for genipin to achieve a similar degree of crosslinking was significantly shorter compared with DHT.
It has been shown that crosslinked gelatin with a slow degradation rate was an ideal candidate for cell growth and tissue regeneration.11,25,38 Tabata and colleagues demonstrated that their GMs degraded slowly when the crosslinking period using DHT was increased.10,24 Other studies have also shown that double crosslinking with genipin gradually stabilizes the degradation rate of gelatin over time.25,39 Therefore, effective crosslinking with a proper incubation time or with multiple crosslinker for the formation of strong covalent binding through hydroxyl group bonding can result in scaffold less susceptible to dissolution.
When evaluating data for (dt – d0/d0) ≤0.6, the value of the diffusional exponent (n) in Equation (1) provides a good indication of the water transport mechanism in test microspheres.26,40 Water transport methods in test microspheres are Fickian diffusion (n = 0.43) and Case-II transport (n = 0.85; Fig. 7). Water movement in test microspheres is regulated by diffusion in the Fickian-diffusion model (glassy state). Fickian diffusion is often characterized by a square-root time dependence on the position of the penetrating diffusion front in a polymer slab. 41 In comparison, the water transport mechanism in the Case-II model is primarily due to polymer chain relaxation in test microspheres, accompanied by a significant volume expansion (rubbery state). 41
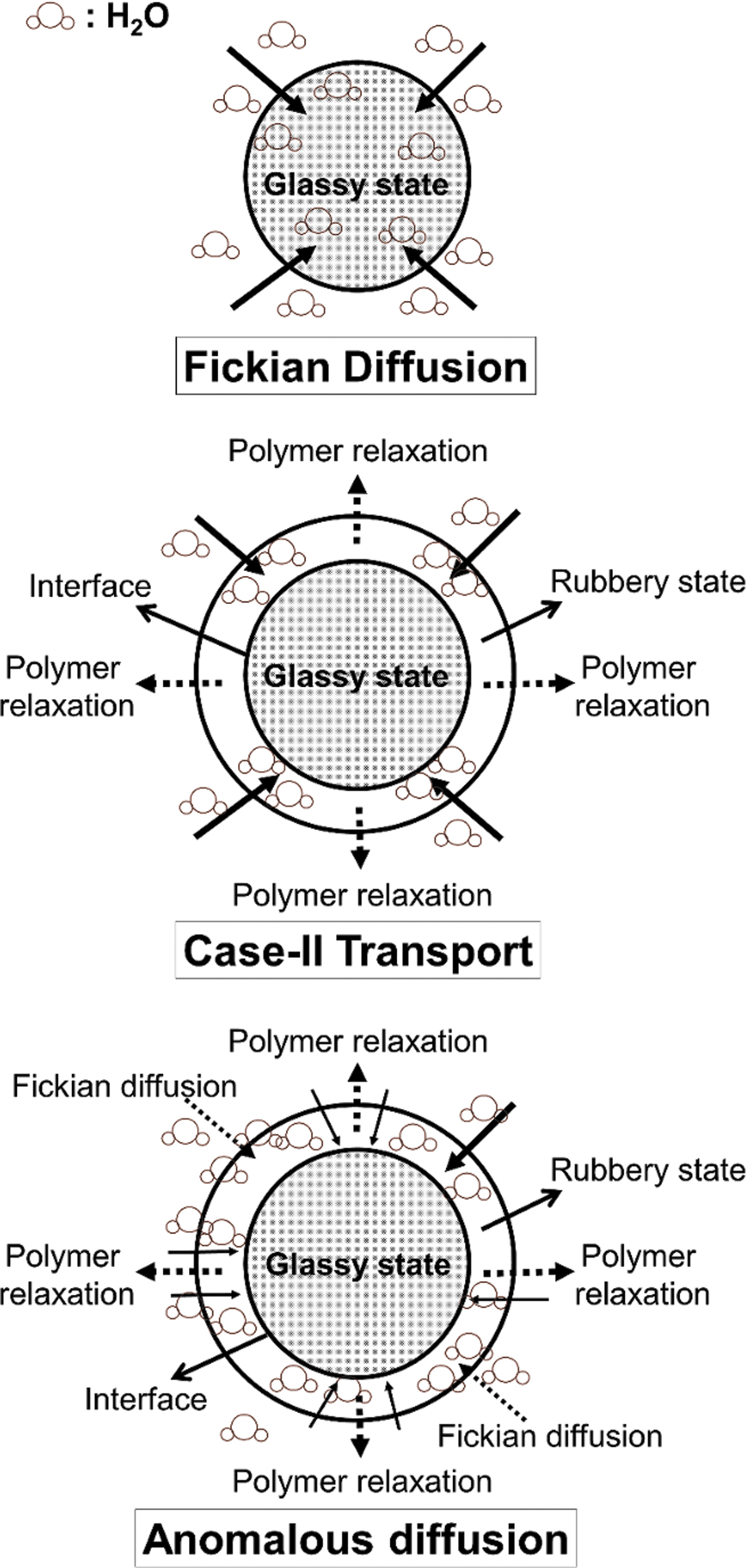
Various concepts for water transfer in test microspheres are illustrated schematically.
The water transport mechanism in test microspheres is said to be anomalous for values of n between these two limits (Fig. 7). When Fickian diffusion and polymer relaxation rates are comparable, this intermediate state exists. In our study, the diffusional exponent (n) in Equation (1) varied between 0.43 and 0.85 for GM-DHT and GM-GEN. As a result, both the GM-DHT and GM-GEN exhibit anomalous water transport mechanisms ranging from Fickian to Case-II extremes (Fig. 7).
Both test microspheres particle sizes increased significantly due to polymer relaxation during immersion in deionized water (Fig. 2G). Therefore, the particle diameter at equilibrium (d∞) for the GM-DHT was slightly smaller than that observed for the GM-GEN (Fig. 1G) without any significant difference in data. In addition, the value of the diffusional exponent (n) in Equation (1) for the GM-DHT (n = 0.47) was smaller than the GM-GEN (n = 0.57). In addition, we compared our data with a few research findings, and discovered that the acquired data were nearly identical to what had been reported.37,40,41
Fluid binding capacity, which is determined by surface chemistry and roughness, is an important feature of scaffolds for tissue regeneration. This information is critical for materials interacting with physiological fluids and cells since it considerably influences structural characteristics and cell–material interactions under in vivo conditions. According to Shankar et al., the scaffolds' time-dependent swelling behavior has a significant impact on determining structural characteristics and cell–material interaction. 42 They discovered that gelatin-crosslinked genipin (G-GEN) has the highest fluid uptake value when compared with two other crosslinked scaffolds (G-DHT and 1,4-butanediol diglycidyl ether).
Although G-DHT had the lowest fluid uptake index, it could absorb more PBS faster than other scaffolds, which can be explained by the material's highest fold increase in fluid-retention ability between 1 h and 7 days of incubation. As shown in this study, gelatin-crosslinked genipin and DHT have demonstrated fluid binding capacity; however, there is no major difference observed between both scaffolds. The fluid uptake findings suggest that the scaffolds were hydrophilic, and able to hold equitable water molecules. The capacity of the material to interact with and retain the water molecule within its network is more dependent on the scaffold microarchitecture.42,43
The study showed the responsiveness of BMSCs on GM-DHT and GM-GEN toward a chondrogenic medium and hypoxic environment, in terms of proliferation, and chondrogenic differentiation is similar to previous findings.44,45 Compared with the limited space in monolayer culture, GM-DHT and GM-GEN provided a greater surface area for cell anchorage and growth. 46 The significant increase in the staining of GM-DHT and GM-GEN chondrogenic differentiation indicated the enhanced effect of GM on BMSC differentiation (Fig. 5C).
However, the mechanism for such effect remains unclear, although some speculated that it could be due to the synergistic effect of increased proliferation of mesenchymal stem cells on GM or the presence of arginine-glycine-aspartic acid sequence on gelatin, mimicking those in the native ECM of chondrocyte.47,48 To ensure its regenerative capabilities, the maintenance of BMSCs stemness properties is paramount. 49 The GM-DHT and GM-GEN maintained BMSCs stemness properties by expressing CD73, CD90, and CD105. Therefore, it is safe to conclude that the BMSCs cultured in GM-DHT and GM-GEN 3D culture are well suited to increase cell expansion.
According to Li et al., cultured cartilage constructs enhanced tissue net weight gain and chondrogenic gene expression. 50 In our investigation, BMSCs-GM-DHT continuously expressed higher levels of anabolic markers such as Sox9, the type I/II collagen ratio, ACAN, and sGAG than monolayer BMSCs-GM-GEN, with the highest levels on day 14. Although the expression of all anabolic markers in proliferating cells is decreased in BMSCs-GM-GEN, it is still significantly higher than that in monolayer culture.
After 14 days of culture, both BMSCs-GM-DHT and BMSCs-GM-GEN demonstrated a decrease in ADAMTS5 and MMP13. This outcome is most likely owing to the proclivity of BMSCs-GM-DHT and BMSCs-GM-GEN to differentiate into the chondrogenic lineage. However, it is important to note that gene expression may also be influenced by cell confluency as previously shown in our study. 11 Nevertheless, taken all the findings together, BMSCs-GM-DHT appeared to be superior as a cell microcarrier for chondrocyte delivery compared with BMSCs-GM-GEN.
One of the study's shortcomings is that BMSCs-GM-DHT and BMSCs-GM-GEN aggregate in culture. Aggregates clog the intra-articular needle and obstruct cell delivery, reducing MSC survival after injection due to shear stress. 51 By enabling continuous rotation of the cell suspension in alternating directions, the dynamic culture of 3D BMSC-GM may circumvent this constraint. 52 The GM would provide a platform for the BMSC to grow and distinguish itself more rapidly without compromising its quality. Simultaneously, the dynamic state would diminish aggregation and increase cell viability.
Conclusion
This study reports a comparative analysis of two coupling agents used to modify GM for cartilage tissue engineering. Upon evaluating the influence of thermal (DHT) and natural (genipin) coupling agents on gelatin crosslinking, we found that the thermal crosslinking process was more appropriate to obtain a 3D scaffold with physical–chemical and biological features. We demonstrated that thermal dehydration of the gelatin improves the overall characteristics without affecting the natural properties of the GM-DHT scaffold. Moreover, the absence of any chemical residues within the protein network would prevent antigenicity in physiological conditions. Similarly, the GM-GEN showed acceptable chemical composition, topography, hydrophilicity, and satisfying biological properties. However, the GM-GEN scaffold's performance was slightly lower compared with GM-DHT. In conclusion, the DHT treatment is the most effective method to obtain promising scaffolds for tissue engineering.
Footnotes
Acknowledgment
The authors thank the National University of Malaysia for providing research facilities and research grants.
Authors' Contributions
S.S., and N.M.H. contributed to conceptualization, methodology, formal analysis, data curation, writing—review and editing; N.M.H. performed validation; S.S. carried out investigation; N.M.H., Y.T., Y.H., R.A.R., and N.H.M.Y. contributed to resources; S.S. contributed to writing—original draft preparation; S.S. performed visualization; N.M.H., Y.T., Y.H., R.A.R., and N.H.M.Y. carried out supervision and project administration; N.M.H. performed funding acquisition. All authors have read and agreed to the published version of the manuscript.
Ethics Approval
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Research Ethics Committee, The National University of Malaysia (UKM PPI/111/8/JEP-2018-458, October 11, 2018). Informed consent statement: informed consent was obtained from all subjects involved in the study.
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by the National University of Malaysia research grant FF-2019-198 and DIP-2015-025.