
Abstract
In the peripheral nervous system, Schwann cells (SCs) play a crucial role in axonal growth, metabolic support of neurons, and the production of myelin sheaths. Expansion of SCs after extraction from human or animal nerves is a long and often low-yielding process. We established a rapid cell culture method using a defined serum-free medium to differentiate human induced pluripotent stem cells (iPSCs) into SCs in only 21 days.
The SC identity was characterized by expression of SRY-Box Transcription factor 10 (SOX10), S100b, glial fibrillary acidic protein (GFAP), P75, growth-associated protein 43 (GAP43), and early growth response 2 (EGR2) markers. The SC purity reached 87% as assessed by flow cytometry using the specific SOX10 marker, and 69% based on S100b expression. When SCs were cocultured with iPSC-derived motor neurons two-dimensionally or three-dimensionally (3D), they also expressed the markers of myelin MBP, MPZ, and gliomedin. Likewise, when they were seeded on the opposite side of a porous collagen sponge from motor neurons in the 3D model, they were able to migrate through it and colocalize with motor axons after 8 weeks of maturation. Moreover, they were shown by transmission electron microscopy to form myelin sheaths around motor axons.
These results suggest that the use of autologous iPSC-derived SCs for clinical applications such as the repair of peripheral nerve damage, the treatment of spinal cord injuries, or for demyelinating diseases could be a valuable option.
Impact Statement
Peripheral nerve injuries can cause the complete paralysis of the upper or lower limbs, which considerably reduces the quality of life of patients. To repair this injury, many approaches have been developed by tissue engineering. Combining biomaterials with Schwann cells (SCs) has been shown to be an effective solution for stimulating nerve regeneration. However, the challenge faced concerns the strategy for obtaining autologous SCs to treat patients. A promising approach is to differentiate them from the patient's own cells, previously induced into pluripotent stem cells. We propose a fast culture method to generate functional SCs differentiated from induced pluripotent stem cells.
Introduction
In the peripheral nervous system, Schwann cells (SCs) originate from neural crest cells that differentiate into SC precursors before reaching the mature SC stage. 1 SCs provide mechanical as well as metabolic support to neurons with the secretion of neurotrophic factors.2–4 They are also responsible for the myelination of axons and the formation of myelin sheaths allowing the faster propagation of nerve impulses.5,6 Trauma, nerve injury, or peripheral demyelinating diseases can affect myelin, SCs, and neurons, leading to degeneration.7,8 Interestingly, SC use has gained much popularity over the years based on their unique ability to enhance axonal migration after an injury.3,4,9–12 Indeed, SCs are now seen as a key cell type to be integrated in any approach aiming to repair peripheral nerve lesions. 13 Moreover, they have been shown to efficiently enhance spinal cord repair while being much easier to generate compared with oligodendrocytes.14,15 Thus, the need to generate autologous SCs for different cell therapies might likely increase in the next decade. 16
SCs can be obtained from a variety of sources. They can be expanded as primary cultures after extraction from animal17,18 or human peripheral nerves.14,19,20 They can also be differentiated from animal or human embryonic stem cells, 21 dental pulp cells, 12 or adipose stem cells. 22 However, primary cultures of SCs require the access to biopsied nerve tissue in the perspective of an autologous clinical application, which may be a major limitation. Moreover, human SC extraction from nerves is a challenging technique to obtain appropriate yields, and cells do not proliferate well in vitro, in contrast to rat SCs. 19 Despite the success of differentiation into SCs of animal or human embryonic, adult, or induced pluripotent stem cells, various hurdles reviewed in 13 are still limiting the translation of these approaches to the clinic. First of all, the SC differentiation and expansion protocols are time consuming, taking between 4 and 8 weeks to get the desired amount of cells.17,19,23,24 In addition, fibroblast contamination has been frequently reported.23–25 Finally, some teams have tried to purify SCs, but the results were at the expense of cell yield.19,24,25
Indeed, the clinical use of primary human SCs remains complex due to time constraints. The use of embryonic and mesenchymal adult stem cells can sometimes raise ethical and safety problems and concerns due to invasive harvesting techniques or slow proliferation. 26 Therefore, studies have turned toward the use of induced pluripotent stem cells (iPSC), which are easier to access, no longer raise ethical concerns, and can rapidly proliferate.27,28 The successful differentiation of hPSCs into SCs was achieved, but over long culture period requiring between 26 and 50 days.4,29,30 However, Kim et al. showed using hPSCs that cells could be frozen at a SC precursor stage, which only require 8 days final step of differentiation after thawing to obtain SCs. 4
Moreover, the culture medium often contain fetal bovine serum (FBS), which is known to promote cell growth but exhibits several inconveniences, such as high batch-to-batch discrepancies. Moreover, due to the risk of animal-derived pathogens' transmission, the use of FBS for a clinical application may be a limiting factor. 31 Now, the use of serum-free culture medium was shown to successfully support rat-derived SC myelination of motor neurons in vitro. 32
The development of an improved protocol enabling the differentiation of SCs from iPSCs in a short period of time (less than a month), with a high yield and a clear demonstration of their functionality (formation of myelin sheaths), is still needed.
We developed a protocol for the differentiation of SCs derived from iPSCs allowing to obtain mature SCs in only 21 days using an FBS-free culture media. We also demonstrated the ability of these iPSC-derived SCs to myelinate motor axons and form myelin sheaths using a three-dimensional (3D) in vitro model to ascertain their functionality.
Materials and Methods
All human cells used in this work were obtained with informed consent from the donors. The study was approved by the CHU de Quebec-Université Laval Research Ethics Review Board.
iPSCs were obtained from the iPS-Quebec platform of the CHU de Québec-Université Laval Research Center. They were derived from three healthy human lymphoblastoid cell lines, 555-25651, 555-617, and 555-1839 and reprogrammed with the CytoTune™-iPS 2.0 Sendai Reprogramming Kit (Invitrogen, Carlsbad, CA).
Extraction of human SCs from a nerve biopsy
The epineurium of a human sural nerve was removed under aseptic conditions with the help of a dissection microscope, by grasping the epineural sheath and pulling on the nerve fascicles with precision forceps. Epineurium-free nerve fascicles were diced into small (2 × 2 mm) pieces and placed in the center of gelatin-coated Petri dishes with 60 μL/cm2 of Dulbecco's modified Eagle's medium (DMEM) containing 30% fetal calf serum supplemented with 100 U/mL penicillin and 25 μg/mL gentamicin. Tissue explants were cultured in an incubator at 37°C and 8% carbon dioxide for roughly 3–4 weeks to induce Wallerian degeneration and deplete the tissue from fibroblast cells. Observations were performed and medium changed every 2–3 days. When a confluent carpet of fibroblast cells had formed, the explants were moved into a new gelatin-coated Petri dish. After 3–4 weeks, the migration of bipolar SCs were observed migrating out from the tissue onto the culture dish.
At this moment, the fascicle explants were collected and digested under gentle movements in a spinner flask for 4 h with a sterile enzymatic dissociation solution containing: DMEM, 1 × Hepes (MP Biomedicals), the addition of 3.1 mM CaCl2 (Sigma), 1.25 U/mL dispase (Roche), and 0.125 U/mL collagenase (Roche). The solution was centrifuged and resuspended in 0.05% trypsin/0.01% ethylenediaminetetraacetic acid (EDTA) for 10 min. After another centrifugation, the cells were seeded onto 10 μg/mL poly-D-lysine and 10 μg/mL laminin-coated culture dishes in SC medium containing 3:1 DMEM/F-12 media, 2% fetal calf serum (HyClone), 1 × N2 supplement (Thermo Fisher Scientific), 2 mM l-alanyl-
Preparation of the collagen/chitosan sponges
Sponges were prepared as previously described (Berthod et al. 1994, 1997)33,34 but without chondroitin 4–6 sulfates. Shortly, types I and III bovine collagen (Symatese, Chaponost, France) and chitosan (Kemestrie, Sherbrooke, Canada) were dissolved in 0.1% acetic acid and mixed. Around 0.5 mL of the mixture was poured into 12-well plates. The frozen plates were then lyophilized in a freeze dryer (Dura-Stop Microprocessor Controlled Tray Freeze Dryer; FTS Systems, Stone Ridge, NY).
Experiment
Differentiation of iPSC into motor neurons
Differentiation of iPSC into motor neurons was conducted following a modified previously published protocol adapted from references.35,36 Briefly, iPSCs were passaged with StemPro Accutase (Thermo Fisher Scientific, Waltham, MA), combined with 5 μM Y-27632 (Abcam, Toronto, Canada), diluted in DMEM-F12 medium without serum. iPSCs were cultured onto Geltrex (1:200; Thermo Fisher Scientific)-coated wells and dissociated at confluency with StemPro Accutase containing 5 μM Y-27632. From day 0 to 11 of differentiation, cell culture medium contained DMEM-F12: neurobasal A medium (vol:vol) (Thermo Fisher Scientific) with 1% N2 Supplement (Thermo Fisher Scientific), 1% B-27 Supplement minus vitamin A (Thermo Fisher Scientific), 0.01 mM ß-mercaptoethanol (Thermo Fisher Scientific), 50 μg/mL ascorbic acid (Millipore Sigma), 5 μM Y-27632, 1% L-alanyl-L-glutamine (Corning, New-York, NY), 1% MEM nonessential amino acids (Thermo Fisher Scientific), 0.1% trace elements A, 0.1% trace elements B, 0.1% trace elements C (Thermo Fisher Scientific), and 100 U/mL of penicillin and 25 μg/mL of gentamicin.
From days 0 to 4 of differentiation, 40 μM SB431542 (Millipore Sigma), 0.2 μM LDN193189 (Millipore Sigma), and 3 μM CHIR99021 (Millipore Sigma) were added to the medium. From day 2 to 9 of differentiation, medium was supplemented with 0.88 nM retinoic acid (Millipore Sigma) and 500 nM SAG (Abcam). From day 9 to 13, for two-dimensional (2D) culture, or to day 19, for 3D culture of differentiation, 10 μM DAPT (Selleck Chemicals LLC, Houston, TX) was added to the medium. At day 11, cells were dissociated with StemPro Accutase containing 5 μM Y-27632 and seeded at 285,000 cells/cm2 over the 3D sponge model or at 200,000 cell/cm2 on 10 μg/mL poly-D-lysine (Millipore Sigma)-coated plates. For 2D culture, from days 11 to 20, differentiation culture medium was the same as the one described previously but β-mercaptoethanol was removed. From day 11 until the end of culture, 20 ng/mL BDNF (Bon Opus Biosciences LLC, Millburn, NJ), and 10 ng/mL GDNF (ABM, Richmond, Canada) were added to the medium.
From day 13 until the end of culture, 10 ng/mL NT-3 (FroggaBio, Concord, Canada) was added to the differentiation medium. For 3D culture, from day 18 until the end of culture, 10 ng/mL NGF was added to the medium.
Differentiation of iPSC into SCs
iPSCs were differentiated into SCs according to a modified protocol from references.36,37 Up to day 3, iPSCs were grown under the same conditions as for motor neuron differentiation (see differentiation of iPSC into motor neurone) to reach a neuronal precursor stage. Cells must be confluent for 3 days. At day 3, cells were cultured in minimum essential medium (MEM, STEMCELL Technologies, Vancouver, Canada) containing 1% N2 supplement, 1 mM β-mercaptoethanol, 5 mM Y-27632, 1% L-alanyl-L-glutamine, 1% MEM nonessential amino acids, 0.1% trace elements A, 0.1% trace elements B, 0.1% trace elements C, and 100 U/mL of penicillin and 25 μg/mL of gentamicin. From day 4, β-mercaptoethanol was removed and 0.88 nM retinoic acid was added until day 6. Cells were passaged at day 5 with StemPro Accutase, supplemented with 5 μM Y-27632, diluted in DMEM-F12 medium added with 5 μM Y-27632 without serum, seeded at 135,000 cell/cm2 on Geltrex (1:200) plates and cultured in differentiation medium added from this day with 1% B-27 supplement without vitamin A.
At day 7, iPSCs were passaged again, seeded under the same conditions as on day 5, and cultured in the medium combined with 5 ng/mL PDGF-bb (Thermo Fisher Scientific), 10 ng/mL βFGF, 14 μM forskolin (Millipore Sigma), and 192 ng/mL NRG1 (R&D System, Oakville, Canada). From day 9, MEM was replaced by a mix 2:1 of DMEM-F12:neurobasal A medium and 1% BSA-20% (Bio Basic, Markham, Canada). Cells were dissociated at day 14 with StemPro Accutase, combined with 5 μM Y-27632, diluted in DMEM-F12 medium added with 5 μM Y-27632 without serum, seeded at 95,000 cell/cm2 on 10 μg/mL poly-D-lysine and 20 μg/mL laminin (Millipore Sigma)-coated plates. At day 21, cells were passaged and seeded at 285,000 cells/cm2 over the 3D sponge model or at 200,000 cells/cm2 for 2D coculture with neurons.
Preparation of the 3D tissue-engineered models
For the 3D coculture model, 800,000 human dermal fibroblasts were seeded onto the collagen/chitosan sponge and cultured for 1 week immersed in DMEM-F12 combined with 10% HyClone serum, 25 μg/mL of penicillin/gentamicin and with 50 μg/mL ascorbic acid. The sponge was flipped and lifted to the air–liquid interface and, 400,000 human epineurium fibroblasts were added and cultured for 1 week with DMEM-F12 supplemented with 10% fetal calf serum and 25 μg/mL of penicillin/gentamicin. Then, the sponge was rinsed with DMEM-F12 to remove any serum residue and one million motor neurons at day 11 of differentiation were seeded on the same side as the dermal fibroblasts. The 3D model was cultured in the same medium than the one used for the motor neuron 2D culture described above for 2 days.
Then, at day 13 of motor neuron differentiation, medium culture was composed of DMEM-F12:neurobasal A medium (vol:vol), 1% N2 Supplement, 1% B-27 Supplement minus vitamin A, 50 μg/mL ascorbic acid, 5 μM Y-27632, 1% L-alanyl-L-glutamine, 1% MEM nonessential amino acids, 0.1% trace elements A, 0.1% trace elements B, 0.1% trace elements C, 10 μM DAPT, 10 ng/mL GDNF, 20 ng/mL BDNF, 10 ng/mL NT-3, and 100 U/mL of penicillin and 25 μg/mL of gentamicin.
One week after the motor neuron seeding, the SCs were added on the opposite side as the motor neurons and the complete model was cultured for an additional 8 weeks in the same medium than for MN.
Quantification of SC purity by flow cytometry
Purity of SCs was quantified by comparing the expression of SOX10 (Recombinant PE Anti-SOX10 antibody [SP267]; Abcam), S100B (Monoclonal Anti-S100B[SH-B1]; Sigma) as primary antibody, and anti-Mouse Alexa Fluor™ 488; Life as secondary antibody) or GAP43 (Recombinant Anti-GAP43 antibody [EP890Y]; Abcam as primary antibody and anti-Rabbit IgG Alexa Fluor 488; Life as secondary antibody) to an isotype antibody (PE Rabbit IgG Isotype Control Antibody [EPR25A]; Abcam) for SOX10 or the control isotype X0931 Mouse IgG1; Dako antibody with anti-Mouse, Alexa Fluor 488 as secondary antibody for S100B, whereas for GAP43 the secondary antibody alone was used as control.
Briefly, cells were fixed 20 min in Paraformaldehyde 4%, blocked and permeabilized 20 min with phosphate-buffered saline (PBS) 0.03% Triton X-100 and 5% FBS and incubated 1 h with the respective antibody in PBS 0.03% Triton X-100 and 5% FBS. Cells were washed and analyzed by flow cytometry in PBS 500 μM EDTA and 2% FBS. Data were acquired using a BD FACSMelody™ flow cytometer (BD Biosciences, Franklin Lakes, NJ) and analyzed with FlowJo™ v10.7 Software (BD Life Sciences). Graphics were illustrated using GraphPad Prism version 9.0.2.
Immunofluorescence
The 2D cells and 3D samples were fixed 20 min and 1 h, respectively, at 4°C with 4% paraformaldehyde and rinsed with cold PBS with 0.3% Triton X-100 (Bio-Rad, Mississauga, Canada). The 3D models were cut into 15 mm2 pieces or into 30 μm cross-sections. Then, cells and 3D substrates were incubated 20 min at room temperature in an immunofluorescence staining buffer containing cold PBS, 0.3% Triton X-100 (Bio-Rad) and 5% serum. They were stained overnight at 4°C with the immunofluorescence staining buffer containing primary antibodies and were washed three times with PBS containing 0.3% Triton X-100. Then, cells and 3D substrates were incubated 1 h at room temperature with secondary antibodies diluted (1:500) in the immunofluorescence staining buffer. Finally, they were rinsed twice with PBS containing 0.3% Triton X-100 and once with PBS before mounting with Fluoromount-G containing DAPI (Electron microscopy Sciences, Hatfield, PA). Imaging was carried out using a LSM700 confocal microscope with Zeiss Axio Imager (Carl Zeiss Microscopy, Jena, Germany).
The primary antibodies used were chicken anti-neurofilament M 160 kDa (1:500; Millipore Sigma), mouse anti-SOX10 (1/500; R&D System), S100B (Monoclonal Anti-S100B[SH-B1]; Sigma), rabbit anti-EGR2 (dilution; Abcam), rabbit anti-GFAP (1:500, Abcam), mouse anti-myelin basic protein (MBP; 1:500; R&D System), and rabbit anti-myelin protein zero (MPZ; 1:100; Abcepta, San Diego, CA).
The secondary antibodies used were Alexa Fluor 647 donkey anti-rabbit (Invitrogen), Alexa Fluor 555 goat anti-mouse (Invitrogen), Alexa Fluor 594 goat anti-rabbit (Invitrogen) and Alexa Fluor 488 donkey anti-chicken (Jackson ImmunoResearch, West Grove, PA).
Transmission electron microscopy
The 3D fresh sponges were cut into 5 mm2 pieces and fixed overnight at room temperature in 2.5% glutaraldehyde. Samples were put in 0.1 M cacodylate buffer and then postfixed with osmium tetroxide solution for 30 min. Regions of interest were identified and embedded in LRWhite™ (London Resin Company Ltd., Theale, Berkshire, United Kingdom). Ultra-thin sections stained with uranyl acetate were observed with a transmission electron microscope (JEOL JEM-1230; Soquelec Ltd., Montréal, Canada). Electron microscopy was performed by the imaging platform of the Université Laval.
Results
The protocol to differentiate iPSCs into SCs was modified to avoid the need to supplement the culture medium with FBS. SCs were differentiated on poly-D-lysine-coated plates and their morphology was assessed after 7 (Fig. 1A) and 21 days of differentiation (Fig. 1B) by phase-contrast microscopy. After 21 days of differentiation, they acquired a bipolar morphology with elongated processes. While they began to express a low amount of S100b, a widely used marker of SC differentiation, at day 7 (Fig. 1C), this expression was shown to be increased at day 21 (Fig. 1D). In addition, GFAP and ERG2, two common markers of SCs, were negative at day 7 (Fig. 1E) but were positively expressed at day 21 along with SOX10, GFAP, and GAP43 (Fig. 1F–H). Then, SCs were seeded onto motor neurons differentiated from the same iPSCs and cultured in 2D for 11 days (Fig. 1I, J). Motor neurons expended neurites characterized by the expression of Neurofilament M (160 kDa), an intermediate filament specific to neurons. SCs were characterized by their expression of EGR2 (Fig. 1I, in white), and GFAP (Fig. 1J, in white) and were seen closely associated with neurites.

Characterization of the SCs cultured in 2D. iPSC-derived SCs were seeded on a poly-D-lysine-coated plate for 2D characterization
To assess the purity of SCs after 21 days of differentiation from iPSCs, the proportion of SOX10-positive cells was quantified by flow cytometry on three different iPSCs (Fig. 2A–C). SC purity was estimated by comparing SOX10 expression to an isotypic antibody control. In addition, the result was compared with SOX10 staining of SCs extracted from human nerves and to undifferentiated iPSCs (Fig. 2A–C). We determined a SC purity of 87.4 ± 2.1% for iPSC-derived SCs compared with 97.9 ± 0.2% for SCs purified from human nerves (Fig. 2G). As a negative control, only 5.3% of nondifferentiated iPSCs expressed SOX10 (Fig. 2C). After 28 days of in vitro maturation, 89.6% ± 6.4 of iPSC-derived SCs expressed SOX10. The same approach was performed using the S100b marker, whose expression is modulated by SOX10 at a later stage.38,39 It showed that 69% of iPSC-derived SCs were found positive for S100b whereas 71% of native human SCs expressed the protein, compared with less than 1% for nondifferentiated iPSCs (Fig. 2D–G).
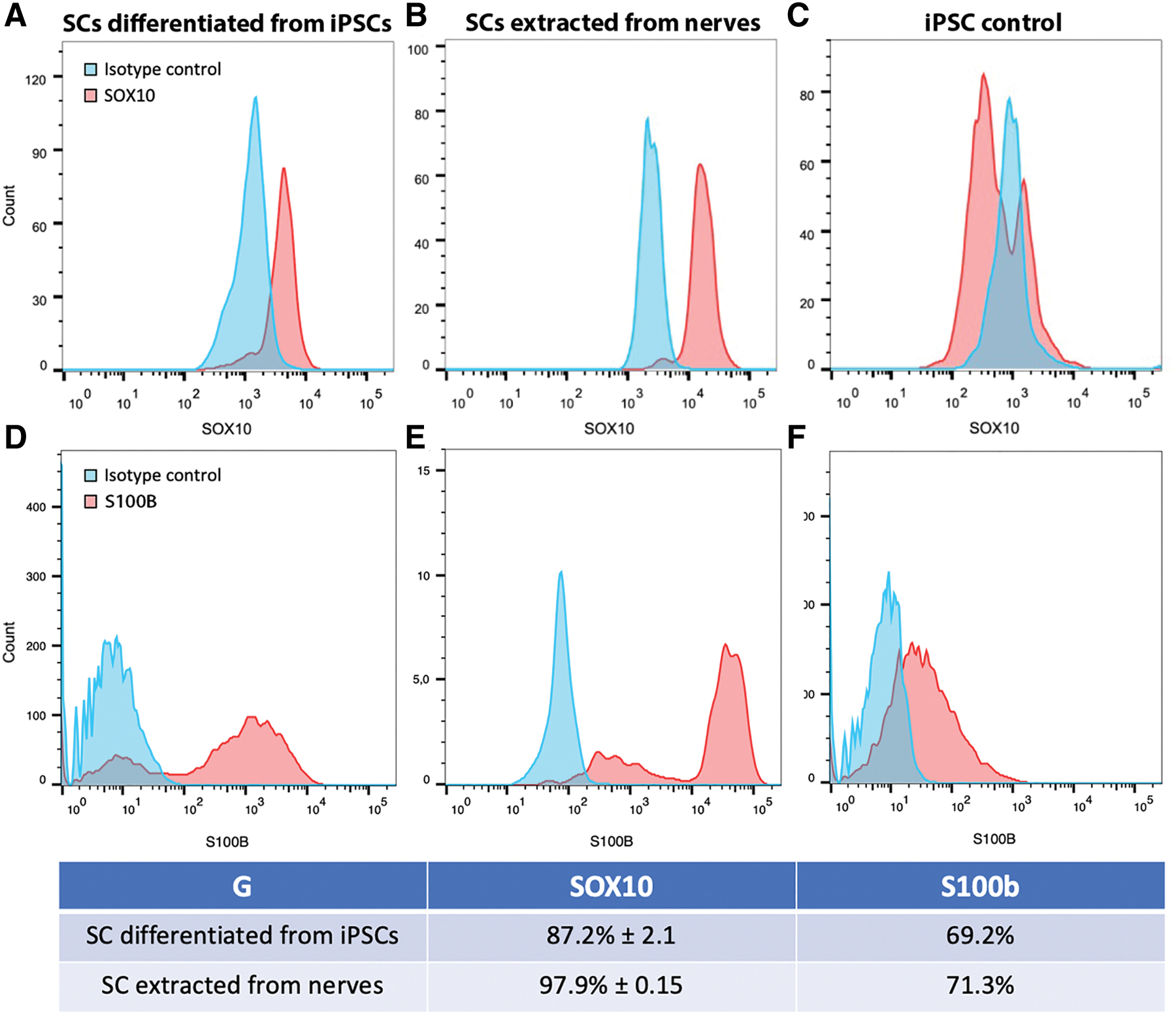
Quantification of SC purity and SOX10 and S100b relative expression by flow cytometry. Purity of SCs was quantified by comparing the expression of SOX10
To investigate the maintenance over time and migration and myelination capabilities of iPSC-derived SCs in a 3D environment, the SCs were differentiated on plastic for 21 days. In parallel, the iPSC-derived motor neurons were differentiated for 11 days on plastic and seeded onto the 3D sponge model where they were matured for 7 additional days. We chose to use iPSC-derived neurons rather than rat or mouse neurons extracted from dorsal root ganglia because there is a very high risk of donor SCs contaminating the neurons during the extraction process. 36 The use of iPSC-derived motor neurons assures that the observed myelination process is indeed due to iPSC-derived SCs, and not to native SCs. Our iPSC-derived SCs were deposited on the opposite side of the sponge as the motor neurons (Figs. 3 and 4). The 3D model was cultured for 8 weeks to induce neurite elongation and SC migration through the substrate and facilitate their association. The interaction of SCs with motor neurites was observed on the side of the sponge where motor neurons were located (view from above) or on transversal cross-sections.

Myelin marker expression in the 3D coculture model. One week after seeding the iPSC-derived motor neurons in the 3D model, the iPSC-derived SCs were added to the opposite side of the sponge and cultured for an additional 8-week maturation. Cells located on the top of the 3D model (where motor neurons were seeded) were imaged from above
Colocalization of glial markers with motor neuron neuritis. One week after seeding the iPSC-derived motor neurons in the 3D model, the iPSC-derived SCs were added to the opposite side of the sponge and cultured for an additional 8-week maturation. Cells located on the top of the 3D model (where motor neurons were seeded) were imaged from above
SCs were still detectable in close proximity from neurofilament M (NFM)-positive motor neurons (Fig. 3A, E, I), as assessed by their expression of SOX10 (B), GFAP (F), P75 (C), gliomedin (G), MBP (J), and MPZ (K). Of note, when undifferentiated iPSCs were added in this 3D model as a control instead of iPSC-derived SCs in the same culture conditions, no staining for SOX10, P75, GFAP, gliomedin, MBP, or MPZ was detected (Data not shown).
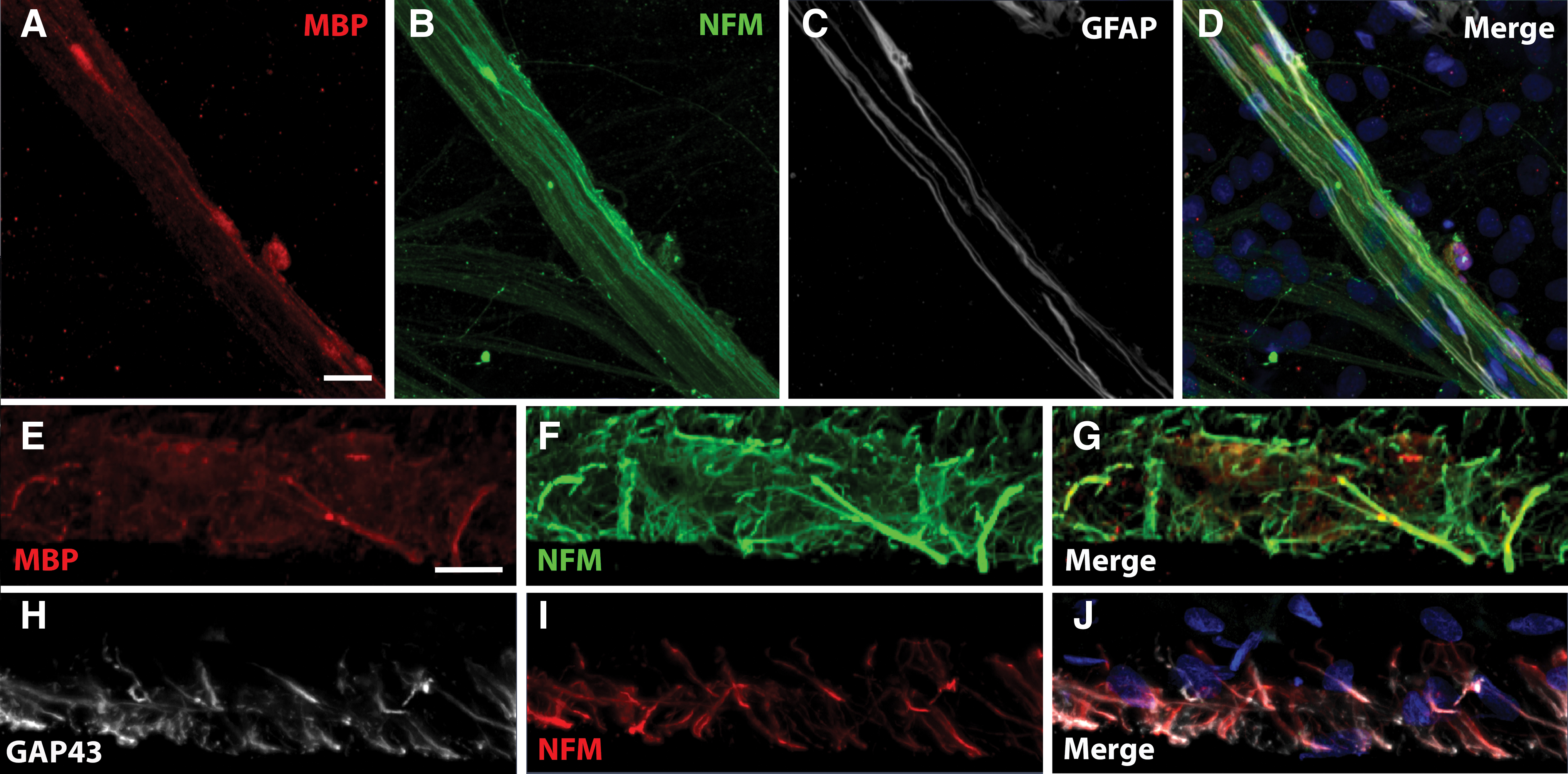
A close interaction was observed between NFM-positive neurites (Fig. 4B) and SCs stained with the glial markers GFAP (Fig. 4C) or MBP (Fig. 4A), from which marker expression increases during SC development.4,40 Such colocalization showed that SCs seeded at the bottom of the model were able to migrate to the top and myelinate neurites. The interaction of SCs with motor neurites was also assessed inside the 3D model on transversal cross-sections (Fig. 4E–J). A similar close interaction between MBP-positive SCs and NFM-positive neurites was observed, mainly on larger caliber neurites (Fig. 4E–G). A colocalization was also observed between GAP-43-positive SCs and NFM-positive neurites (Fig. 4H–J).
To confirm the ability of SCs to form myelin sheaths around neurites, the 3D samples were observed by transmission electron microscopy on ultra-thin tissue cross-sections. After 8 weeks of in vitro maturation, several myelin sheaths enwrapped around neurites were observed with a sheath thickness of ∼0.2 μm (Fig. 5A–C).

Formation of myelin sheaths around the motor neuritis. Myelin sheaths were observed by transmission electron microscopy in the 3D model after coculture of iPSC-derived SC with iPSC-derived motor neurons for 8 weeks
Discussion
The purpose of this study was to develop a faster method to differentiate human-derived iPSCs into SCs than previously achieved in the literature while also obtaining a high differentiation yield in the absence of fetal calf serum. Moreover, it was mandatory to show that these cells were able to form myelin sheaths in contact with neurons. As shown by Huang et al., several studies have focused on the differentiation of iPSCs into SCs, and the protocols used differed in duration and media composition.3,4,13,30,41–43 In this study, iPSCs were directly induced into SCs with a protocol adapted from the literature.34,35
Compared with other studies, one of the advantages of our protocol is that we do not need to use bovine serum in either the SC differentiation protocol or the coculture with neurons for the myelin study, thus limiting the use of compounds from animal origin that may be a limitation in the perspective for a clinical application.4,43,44 As early as day 21 of culture, the differentiated cells expressed known SC markers such as S100b, EGR2, GFAP, GAP43, or SOX10, which is often associated with earlier stages but is also necessary for the maintenance of SC identity. 45 The presence of these characteristic markers correlates with several research articles in the literature.3,4,36,41,44
Moreover, our protocol resulted in mature SCs more rapidly than other currently available protocols, which often require 30 days or more of culture.3,4,30,41 In addition, many studies require a first stage of differentiation as embryoid bodies or neurospheres,3,30,41,43 whereas our method is oriented through a neuronal commitment before further differentiation into SCs.4,42,44 Also, other approaches aimed to develop protocols for obtaining SCs after spontaneous induction, resulting in lower controllability of SC yield, while inducing the formation of other cell types that may be unwanted.42,43
One major advantage of our method is to enable a very high differentiation yield of 87% after 21 days of maturation. We compared this yield with the one achieved after extraction of SCs from human nerve biopsies that reaches 97%. However, it took up to 1 month to obtain these native SCs, and necessitated harvesting a small piece of nerve, which is not always possible and causes morbidity to the patient. In addition, human nerve-derived SCs display a low proliferation potential after extraction. In contrast, the production of SCs by differentiation from iPSCs with such a high yield, allows to provide as many autologous cells as needed without causing any morbidity to the patient (iPSCs were induced from blood-derived lymphoblastoid cells).
To assess the ability of SCs to associate with axons and form myelin sheaths in a 3D environment, we established a model based on a previous coculture experiment between SCs and sensory neurons. 36 Indeed, we were able to show a close association between iPSC-derived SCs and iPSC-derived motor neurons in vitro in conventional 2D cocultures. However, since these SCs are intended to be used to promote nerve regeneration in vivo, it was essential to show their ability to migrate in a 3D environment and spontaneously associate with axons. In this study, we used motor neurons differentiated from iPSCs, instead of extracting them from mouse embryos as previously shown to prevent contamination with endogenous mouse SCs. 18
When SCs were cultured on a collagen sponge previously populated with motor neurons for 8 weeks, they still expressed the glial markers SOX10, P75, and GFAP. In addition, whereas they were seeded on the opposite side of the sponge as motor neurons, we showed that SCs were able to migrate through the tissue to myelinate motor neurites. Once in close contact with neurites, SCs expressed gliomedin, a glial ligand for neurofascin and NrCam associated with nodes of Ranvier assembly in axons.46,47
In addition, they expressed MBP and MPZ, markers of myelin, in regions closely associated with NFM-positive motor neurons or neurites. A staining with MBP and MPZ has been used in various studies assessing the myelinating ability of SCs differentiated from iPSCs4,41,44 or primary cultures purified from mouse nerves. 18 However, it should be noted that staining with gliomedin, MBP, and MPZ were not organized into sheaths regularly interrupted by nodes of Ranvier such as in axons in vivo, probably because of an incomplete in vitro maturation of the nerve fibers.
The ultimate demonstration of the ability of SCs to form myelin sheaths was shown on transmission electron microscopy tissue cross-sections where multilayered myelin sheaths were observed. They displayed a thickness of ∼0.2 μm, which is similar to myelin sheaths formed in vitro by primary cultured animal SCs.17,18,48
In summary, we have developed a serum-free protocol allowing for the direct differentiation of iPSCs into SCs with myelinating capabilities after only 21 days of culture and a purity of 87%. Moreover, these SCs can be frozen at the end of the 21 days of differentiation and used later. Obtaining autologous SCs for a clinical application to repair peripheral nerves, spinal cord, or to treat demyelinating disease represents a real challenge. This approach holds promise for rapidly generating large numbers of highly pure and functionally active autologous SCs. Thus, further investigations would be required to validate their clinical usefulness, including a comparison of genome similarities, secretome, cell marker expression, and in vivo myelinating properties of iPSC-derived SCs compared with cultured human primary SCs.
Footnotes
Authors' Contributions
A.L. and M.-J.B. contributed to the conception and design of the study, acquisition, analysis and interpretation of data, and wrote the article. R.P. contributed to the acquisition, and analysis and interpretation of data. F.B. contributed to the conception and design of the study, analysis and interpretation of the data, and to the article revision and approval for submission.
Ethics Statement
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to, and the approval from the Institutional Review Board of the CHU de Quebec-Université Laval has been received (IRB No. 2019-4444), including informed consent from the patients.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Ataxia Charlevoix-Saguenay Foundation, the National Ataxia Foundation and the Quebec Cell, Tissue, and Gene Therapy Network–ThéCell (a thematic network supported by the Fonds de recherche du Québec - Santé).