
Abstract
The purpose of this study was to document patterns of oligodendrocyte vulnerability to traumatic brain injury (TBI) and determine whether post-traumatic hypothermia prevents oligodendrocyte cell loss. Sprague-Dawley rats underwent moderate fluid percussion brain injury. Thirty minutes after TBI, brain temperature was reduced to 33°C for 4 hours or maintained at normothermic levels (37°C). Animals were perfusion-fixed for quantitative immunohistochemical analysis at 3 (n = 9) or 7 (n = 9) days post-TBI. Within the cerebral cortex, external capsule, and corpus callosum, numbers of APC-CC1 immunoreactive oligodendrocytes at 3 and 7 days following TBI were significantly decreased compared with sham-operated rats (p < 0.02). Double-labeling studies showed that vulnerable oligodendrocytes expressed increased Caspase 3 activation compared with sham. Post-traumatic hypothermia significantly reduced the number of CC1-positive oligodendrocytes lost after normothermia TBI in white matter tracts (p < 0.01). This model of TBI leads to quantifiable regional patterns of oligodendrocyte vulnerability. Post-traumatic hypothermia protects oligodendrocytes by interfering with Caspase 3-mediated cell death mechanisms. Therapeutic hypothermia may improve functional outcome by attenuating trauma-induced oligodendrocyte cell death, subsequent demyelination, and circuit dysfunction.
Introduction
The progressive nature of gray and white matter vulnerability after TBI (Pierce et al., 1998; Dixon et al., 1999; Bramlett and Dietrich, 2002) has been emphasized in experimental and clinical literature. Bramlett et al. (1997b) reported widespread evidence of atrophy within cortical and subcortical areas months after fluid percussion (FP) brain injury. Evidence of severe white matter atrophy and widespread demyelination (Bramlett and Dietrich, 2002) were also documented 1 year post-trauma. Atrophy of white matter tracts and subsequent demyelination is a common occurrence in patients with chronic or multiple TBI (Cullum and Bigler, 1986; Anderson and Bigler, 1995; Gale et al., 1995; Vespa et al., 2010). Although several studies have reported that post-traumatic hypothermia protects against traumatic axonal injury (Koizumi and Povlishock, 1998; Buki et al., 1999), no studies have characterized the effects of post-traumatic hypothermia on oligodendrocyte vulnerability after TBI. Thus, the purpose of this investigation was to assess regional patterns of oligodendrocyte cell death using specific markers for mature oligodendrocytes (Bhat et al., 1996; Fuss et al., 2000; McDonald et al., 2002). We also assessed whether oligodendrocyte vulnerability was associated with Caspase 3 activation. Finally, the efficacy of early post-traumatic cooling on oligodendrocyte vulnerability was investigated. To achieve these goals, we utilized a cooling strategy that has previously been shown to reduce contusion volume and preserve vulnerable populations of neurons (Suzuki et al., 2003). These studies emphasize that post-traumatic hypothermia has a protective effect on oligodendrocyte cell survival in this model of TBI.
Materials and Methods
FP brain injury
Male Sprague-Dawley rats (220 to 270 g; Charles River Laboratories) received TBI using the moderate parasagittal FP injury model that results in neuropathological and behavioral consequence that have been reported to be improved by post-traumatic hypothermia (Dietrich et al., 1994; Bramlett et al., 1995). All experimental procedures were in compliance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the University of Miami Animal Care and Use Committee. One day before TBI, sham (n = 4) and TBI (n = 18) animals were anesthetized with 3% isoflurane, 70% N2O, and 30% O2 and received a 4.8-mm craniotomy (3.8 mm posterior to bregma, 2.5 mm lateral to the midline) to anchor a modified plastic 18-gauge syringe hub (8 mm length, precision guide needle; Becton Dickinson) over the exposed dura of the right parietal cortex. Twenty-four hours after the craniotomy, animals were anesthetized with 3% isoflurane, 70% N2O, and 30% O2, then intubated endotracheally, and mechanically ventilated (Harvard Apparatus) with 1.5% isoflurane, 70% N2O, and 30% O2. To facilitate mechanical ventilation, pancuronium bromide (0.5 mg/kg) was intravenously administered through the femoral artery to immobilize the animals. To ensure consistent physiologic responses among animals, the femoral artery was cannulated to monitor blood gases (PO2 and PCO2), blood pH, and mean arterial blood pressure (MABP), which were maintained within normal physiological range at 15 minutes before TBI and up to 4 hours after TBI.
Sham and TBI animals were attached to the FP device and the TBI animals received a moderate FP pulse (2.0 ± 0.2 atmospheres) delivered to the right parietal cortex. Sham animals underwent all surgical procedures except for the FP pulse. Rectal and temporalis muscle thermistors measured core and brain temperatures, respectively, using self-adjusting feedback warming lamps. Moderate hypothermia was induced by blowing cold air over the rat's head starting 30 minutes after TBI to maintain a brain temperature between 33.0°C and 33.6°C. Sham animals received the identical hypothermia treatment as the TBI hypothermia animals. Normothermia animals were maintained at a brain temperature of 36.6°C to 37.2°C. After TBI or sham injury, hypothermia was maintained for 4 hours followed by a slow rewarming period without heating lamps as previously described (45 minutes to 2 hours) (Suzuki et al., 2003).
Immunohistochemical analysis
At 3 (n = 14) or 7 days (n = 8) after TBI or sham procedures, animals were anesthetized using 3% isoflurane, 70% N2O, and 30% O2 and transcardially perfused with saline (2 minutes, 4°C, 75 mL) and then with 4% paraformaldehyde and phosphate-buffered saline (PBS; 30 minutes, 4°C, 350 mL). The brains were sectioned in PBS (60 μm thick) with a Leica Vibratome (Leica Microsystems, Inc.) from bregma levels − 2.3 mm to − 5.8 mm. Free-floating sections were blocked for 1 hour at room temperature in blocking buffer (PBS containing 5% normal goat serum, and 0.3% TX-100). Sections were then incubated overnight in 4°C in blocking buffer containing primary antibodies, rinsed with PBS, and incubated for 2 hours at room temperature in blocking buffer containing secondary antibodies. The sections were then rinsed with PBS and mounted using Prolong Gold Anti-Fade Mounting Medium (Invitrogen). Primary antibodies used were mouse anti-APC-CC1, against adenomatous polyposis coli gene, mIgG (1:500; Calbiochem) (Bhat et al., 1996; Fuss et al., 2000; McDonald et al., 2002), activated form of rabbit anti-Caspase 3 (1:500; Millipore; AB3623) and rabbit GFAP (1:500; Millipore; AB5804). Controls lacking the primary antibody were run in parallel. Secondary antibodies used were Alexa 488-, 546-, and 647-labeled anti-rabbit and anti-mouse antibodies (Invitrogen). TO-PRO-3 as a nuclear marker (1:2000; Invitrogen) was added with the secondary antibodies or at the end of staining.
Regions of interest, including the peri-contusional cerebral cortex, external capsule, and corpus callosum, were chosen for quantitative analysis using a nonbiased stereological approach to cell counting (Suzuki et al., 2004). Images were obtained by montaging Y-field epiflourescent 40 × magnification images using Neurolucida 7.5.1 software (Micro-Bright Field, Inc.). Higher magnification optical imaging was performed with an LSM510 laser scanning confocal microscope (Carl Zeiss, Inc.) using 25 × 0.8 ma and 63 × 1.2 ma water-immersion lenses. To minimize experimental variability, sections from each experimental group were processed in parallel and imaged during the same imaging session using identical microscope settings and the final images were scaled identically. Sections were selected from five different bregma levels (−2.8, − 3.3, − 3.8, − 4.3, and − 4.8 mm) from each animal in each experimental group and all animals from each group yielded similar results.
Stereology
The number of APC-CC1-positive cells was quantified in the peri-contusional cerebral cortex, external capsule, and corpus callosum. Serial vibratome sections (60 μm) of the rat brain at the level of injury were divided into four groups; each group contained five sections, representing areas containing the contusion as well as regions rostral and caudal. For estimation of the number of APC-CC1 immunopositive cells, diaminobenzidine (DAB) was used as the chromagen. Both cerebral cortex and white matter tract regions in normothermia and hypothermia animals at 3 and 7 days after TBI were analyzed using an Axiophot (Zeiss, Inc.) research microscope, furnished with a fully motorized 3-D LEP stage, Optronix cooled video camera, and MicroBrightField Inc. Stereo-Investigator software package. To perform cell number estimation in the structure, the optical fractionator method and optical dissector probe were used. Dimension of the optical dissector was designed based upon the cell distribution on the section, and optical fractionator grid size was determined based upon the results of the preliminary count of the naïve brain sample to allow 200 counts per brain region. Cortex and white matter tracts were analyzed separately. Immunoreactive cells were those that had degrees of cell body stain greater than controls lacking primary antibody. In addition, positive oligodendrocytes were identified by their morphology as ameboid in shape compared with any potential cross-reactivity with CC1-positive astrocytes as previously described (Bhat et al., 1996). The nonbiased stereological approaches used in this study would be expected to minimize the influence of edema formation and tissue swelling after injury.
Statistical analysis
Data presented are mean ± SEM. Physiological and cell count data are analyzed using one-way analysis of variance followed by post hoc analysis (Fisher LSD) to determine group differences.
Results
Physiological variables
All physiological variables were within normal ranges (pH 7.3–7.4; PO2 121–153 mmHg; PCO2 38–42 mmHg; MABP 105–130) before and following the traumatic insult. Significant differences were seen between brain temperature recordings within the normothermia (36.7°C ± 0.1°C) and hypothermia (33.0°C ± 0.1°C) groups. No other significant differences were seen between any of the experimental groups.
Immunocytochemistry
Immunohistochemical examination of sham-operated animals showed a high frequency of CC1 immunoreactive cells within both gray and white matter structures. Within the cerebral cortex, CC1 immunoreactive oligodendrocytes were scattered within cortical regions with increased density in the lower cortical layers. White matter tracts including the corpus callosum and external capsule contain CC1 immunoreactive oligodendrocytes (Fig. 1A–C). Frequently, these cells were lined up in rows parallel to the projecting axon fibers.
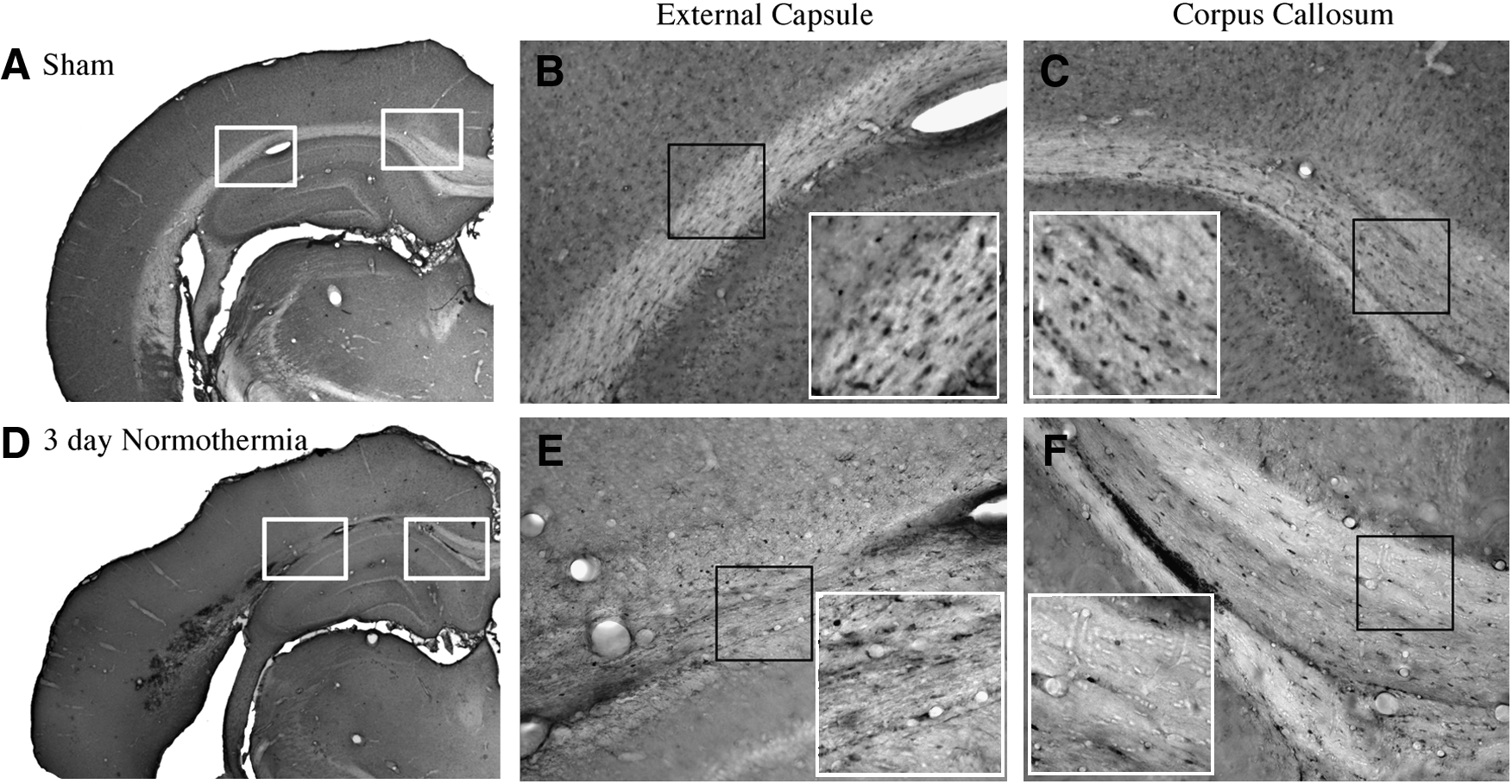
Micrographs of CC1 immunoreactive oligodendrocytes. Sham
At 3 and 7 days after TBI, there was an observable decrease in the number of immunoreactive oligodendrocytes within the lateral parietal cerebral cortex, an area that shows selective neuronal damage in this model (Dietrich et al., 1994), as well as specific areas of the external capsule (Fig. 1D–F). For example, a lack of stained cells within the evolving contusion site was commonly seen (Fig. 1D, E). Within the cortex in adjacent areas, oligodendrocytes appeared reduced in density compared with the contralateral hemisphere. An observable reduction in the frequency of immunoreactive cells was seen in the corpus callosum compared with sham-operated animals (Fig. 1D, F).
Following post-traumatic hypothermia, qualitative assessment of animals at 3 (Fig. 2C, D) and 7 days after injury shows an increase of CC1-positive cells in the pericontusional cerebral cortex and external capsule compared with normothermia animals (Fig. 2A, B).
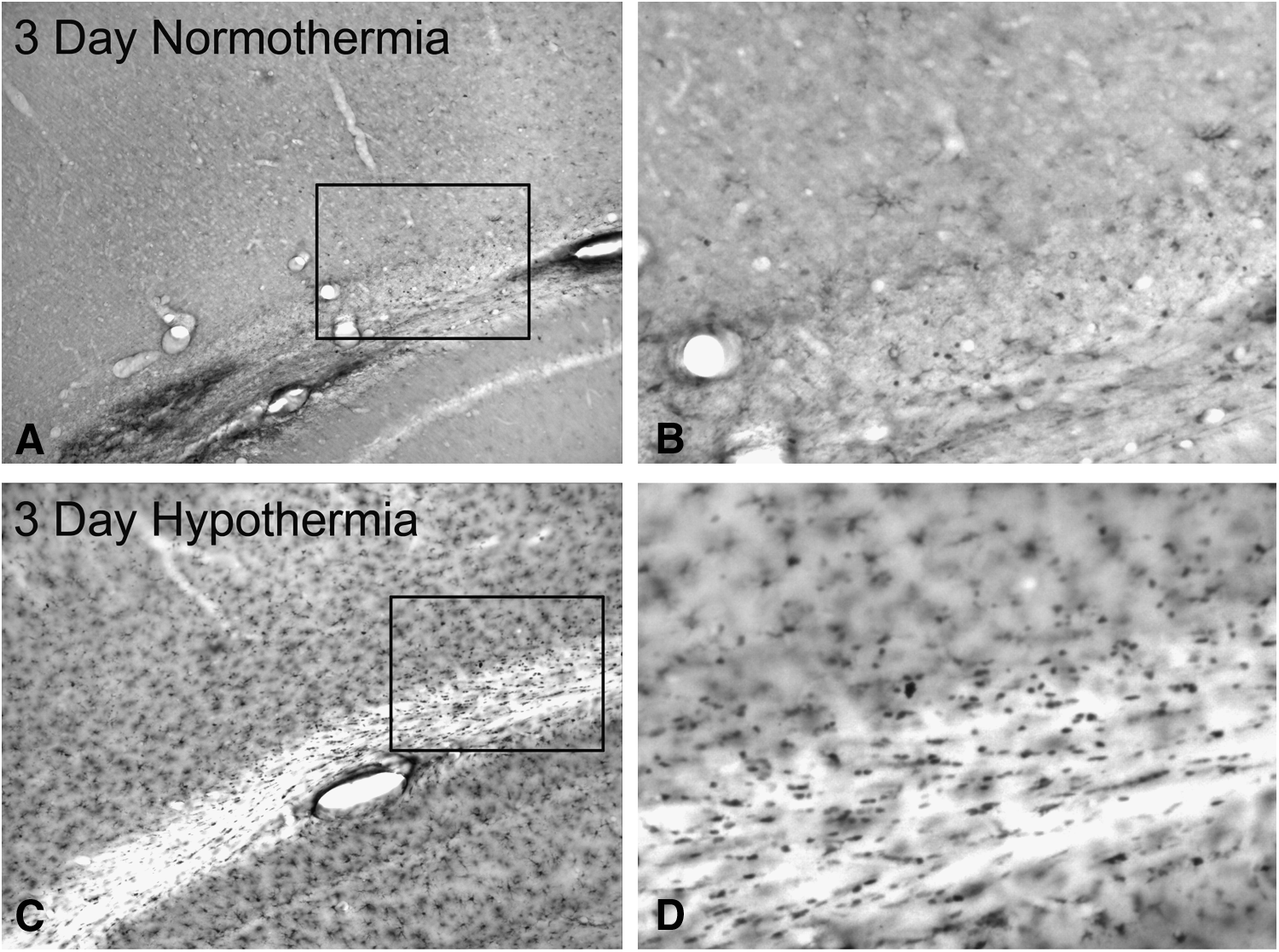
Temperature-dependent changes in CC1 immunoreactive oligodendrocytes.
Previous reports (Bhat et al., 1996; Fuss et al., 2000) utilizing CC1 as an immunomarker for oligodendrocytes have reported that CC1 clearly stains oligodendrocytes and may only weakly stain other cell types such as astrocytes and Schwann cells. Figure 3 demonstrates in our TBI model a lack of double-stained GFAP-positive astrocytes within the pericontusional cerebral cortex, corpus callosum, and external capsule. Therefore, the use of CC1 as a marker for oligodendrocytes and established morphological parameters enabled us to clearly identify oligodendrocytes for quantitative analysis.
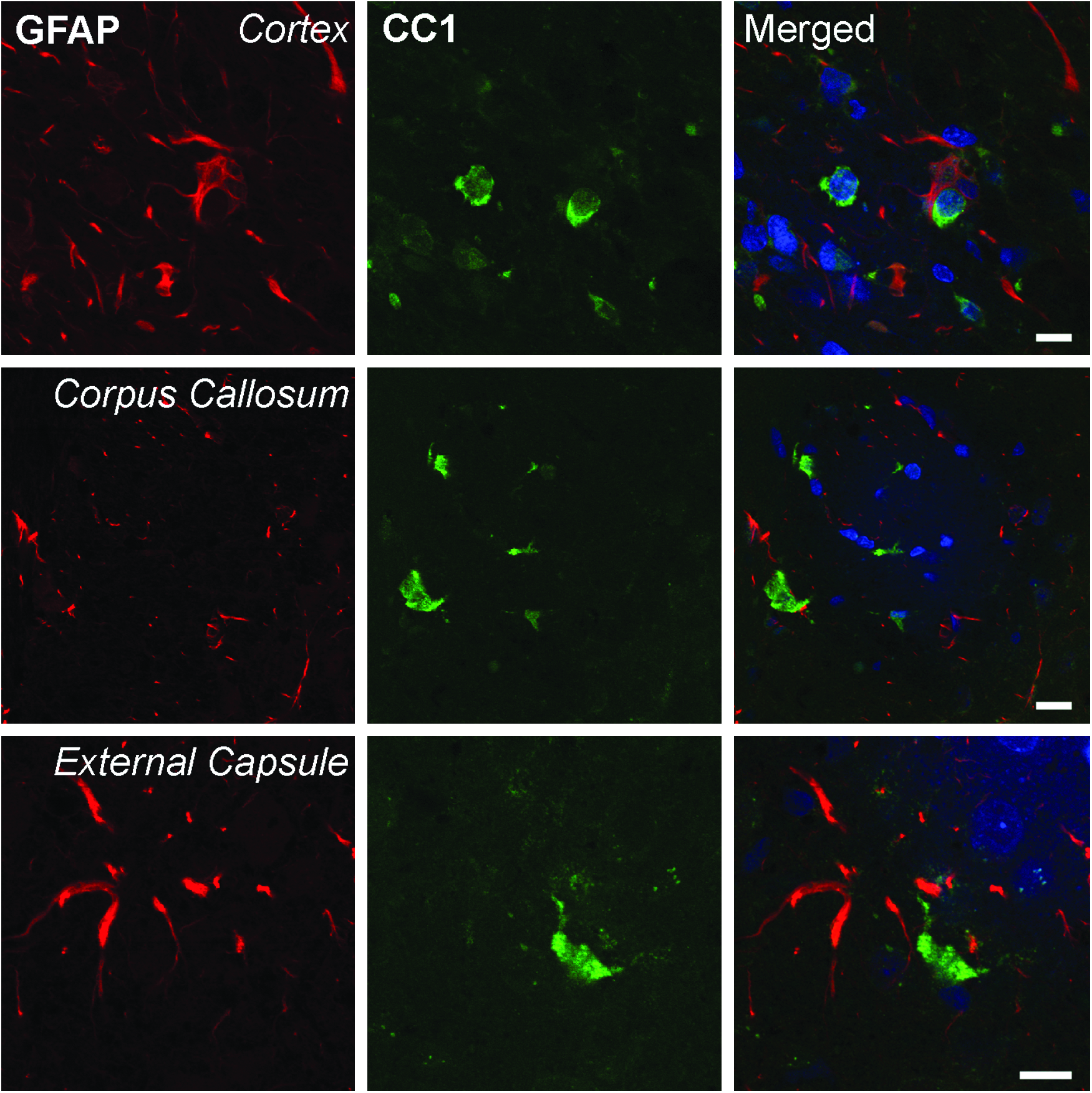
Representative confocal micrographs of double-labeled CC1 and GFAP immunoreactive cells. CC1 (green) and GFAP (red)-positive cells in the cortex (row 1), corpus callosum (row 2), and external capsule (row 3) were examined in traumatized normothermia rats 3 days postinjury. CC1 and GFAP colocalization was not seen in these regions of the rat brain (merged). TO-PRO 3 (blue) was used as a nuclear marker. Scale bars = 10 μm.
Caspase 3 immunoreactivity
Sections were also double-stained with APC-CC1 and activated Caspase 3. A high frequency of double-labeled cells were seen within the cerebral cortex at 3 and 7 days after TBI (Fig. 4). These double-stained cells were scattered among cells that were only CC1 immunoreactive. Similar double-labeling findings were not seen in sham-operated control animals. In addition, post-traumatic hypothermia showed an apparent decrease in double-labeled Caspase 3/CC1-positive cells compared with specimens obtained from normothermia TBI rats. Ongoing studies in the laboratory are quantitatively assessing the frequency of Caspase 3 immunoreactive oligodendrocytes.
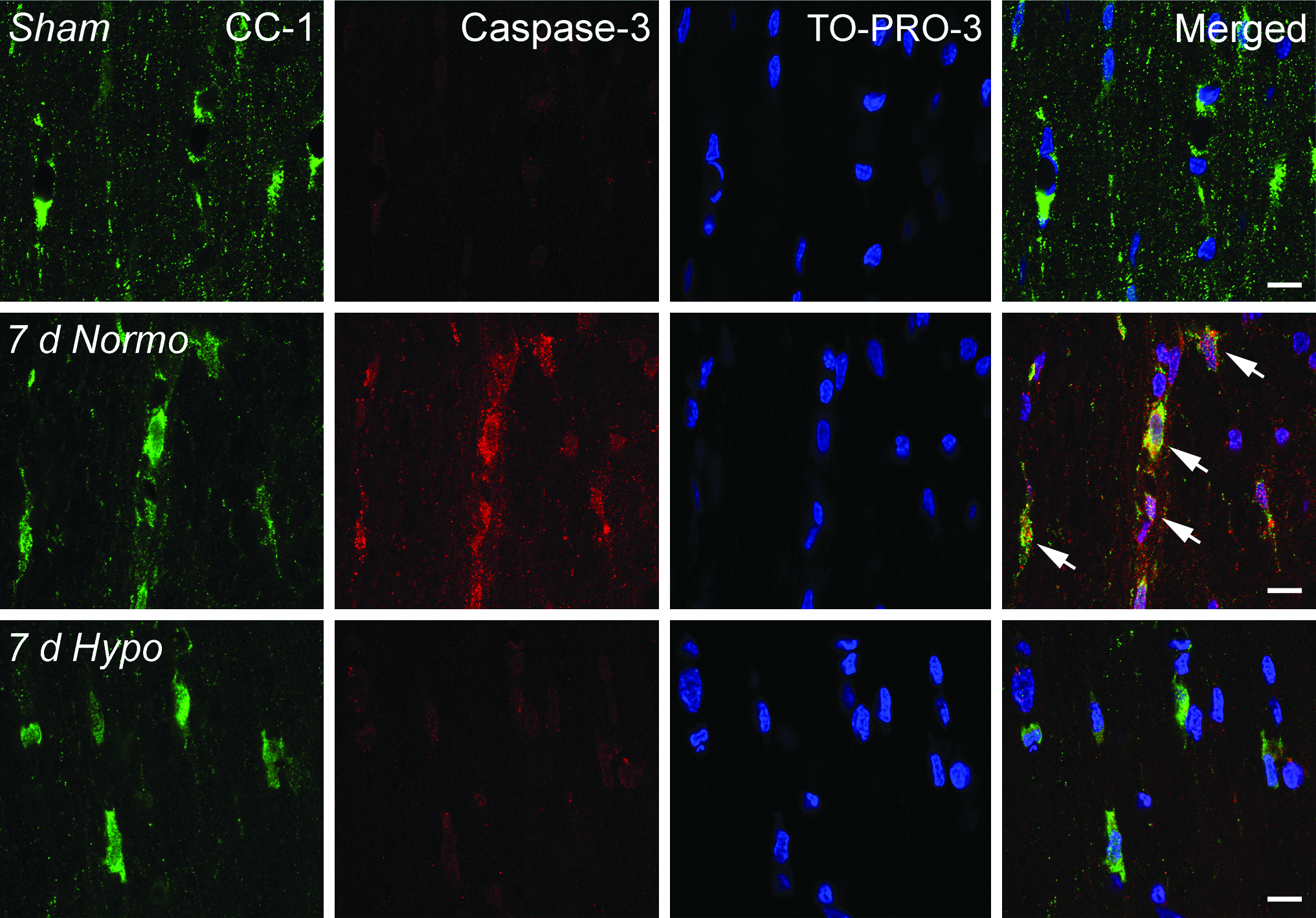
Representative confocal micrographs of double-labeled CC1/Caspase 3-positive cells in the corpus callosum. Sham animals demonstrated CC1-positive cells with no Caspase 3-positive cells (row 1). In contrast, a 7 day normothermia TBI animal exhibits robust double-labeling of Caspase 3 and CC1 (arrows), indicating apoptotic cell death of this oligodendrocyte (row 2). Acute hypothermia treatment appears to reduce the appearance of double-labeled Caspase 3/CC1-positive cells at 7 days after injury (row 3). TO-PRO-3 (blue) was used as a nuclear marker. Scale bars = 10 μm.
Quantitative analysis
Stereological cell counting approaches in sham-operated animals showed consistent numbers of immunoreactive oligodendrocytes within the cerebral cortex, external capsule, and corpus callosum. At both 3 and 7 days after TBI with normothermia, significant reductions in the number of immunoreactive oligodendrocytes were seen in all structures analyzed (Fig. 5). One-way analysis of variance was significant overall for group (p < 0.02). For example, within the lateral parietal cerebral cortex, a dramatic reduction in CC1-positive cells was seen at both 3 (p < 0.003) and 7 (p < 0.02) days after injury compared with sham (Fig. 5B). Similar reductions in immunoreactive cell numbers were also seen within the external capsule (Fig. 5C) and corpus callosum (Fig. 5D). One-way ANOVA was significant for group for the external capsule (p < 0.001) and corpus callosum (p < 0.001). Post hoc analysis again demonstrated significant reductions (p < 0.001) in numbers of CC1-positive cells at 3 and 7 days post-TBI in all groups for the external capsule. Both 3 and 7 day TBI animals demonstrated significant (p < 0.001) loss of CC1-positive cells compared with sham in the corpus callosum.
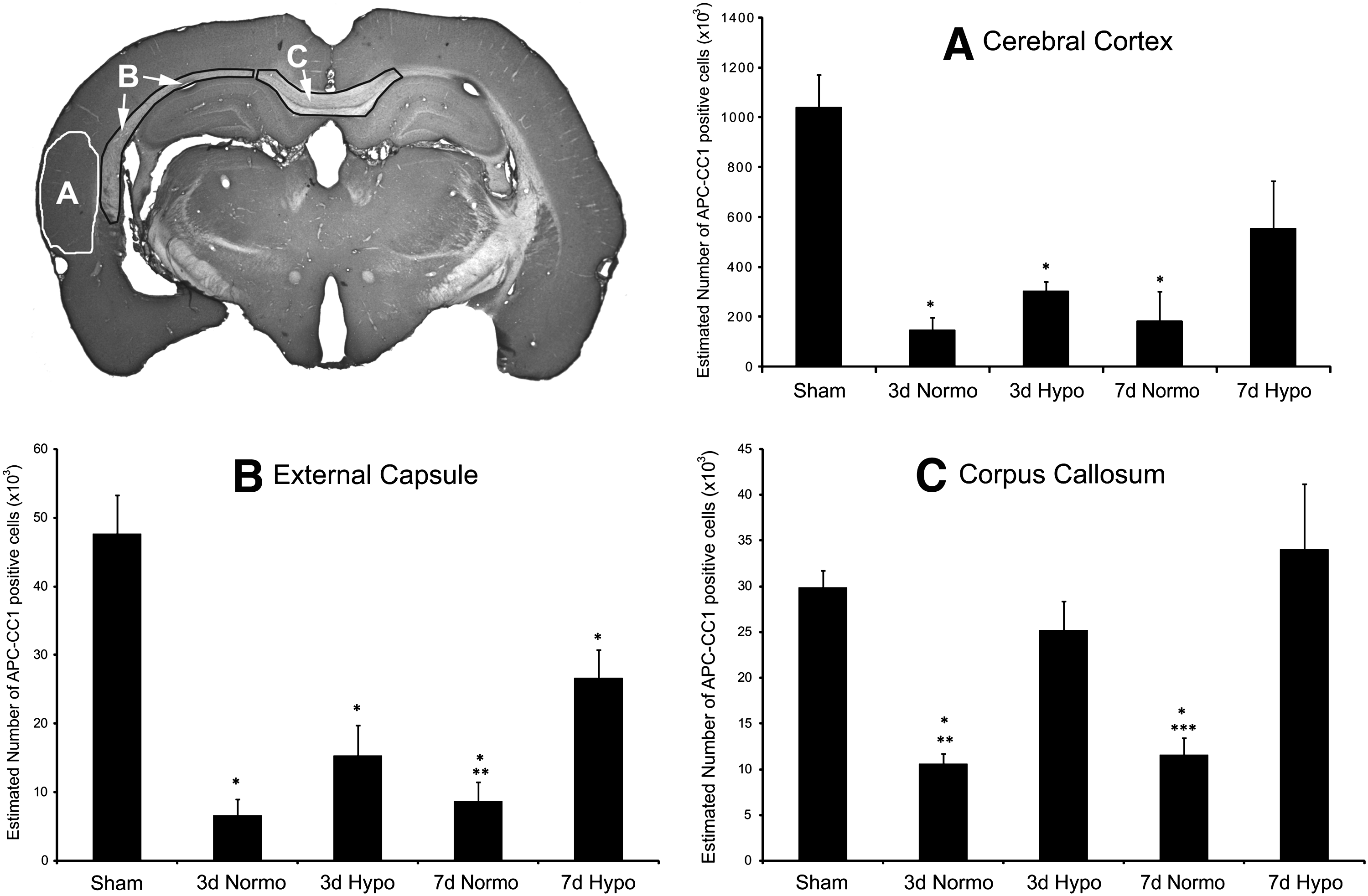
Quantitative assessment of CC1-positive cells within vulnerable gray and white matter regions. Representative micrograph indicating where the stereological cell counts were conducted.
Post-traumatic hypothermia resulted in a significant reduction in the loss of immunoreactive oligodendrocytes in the various brain regions compared with normothermia groups (Fig. 5). At 3 days after injury, a significant (p < 0.005) number of CC1-positive cells were not lost within the corpus callosum following hypothermia compared with normothermia TBI. There was also a trend in the hypothermia groups to reduce the number of lost CC1-positive cells following normothermic TBI in the external capsule and cerebral cortex. At 7 days post-TBI, numbers of CC1-positive cells were significantly increased in both the external capsule (p < 0.004) and corpus callosum (p < 0.001) compared with normothermia TBI animals. Again, there was a trend for the number of cortical-positive CC1 cells to be increased with hypothermia at 7 days compared with normothermia (p = 0.075).
Discussion
Experimental TBI can lead to a host of cellular events that are felt to participate in the long-term functional consequences of brain injury (for review see Bramlett and Dietrich, 2007). In previous studies, significant attention has been directed to patterns of neuronal cell death and diffuse/traumatic axonal injury (Povlishock and Christman, 1995; Bramlett et al., 1997b). Our present findings emphasize that oligodendrocytes, the myelin producing cells of the central nervous system, are also highly vulnerable to moderate TBI. Regional patterns of reduced oligodendrocyte numbers were documented in specific white matter structures within the injured brain. Antibodies specific to oligodendrocytes were used in combination with nonbiased sterological counting procedures that showed significant reductions in oligodendrocyte numbers at two time points after TBI compared with sham-operated control animals (Bhat et al., 1996; Fuss et al., 2000). Double-labeling studies demonstrated that Caspase 3-mediated cell death may participate in the vulnerability of oligodendrocytes to TBI. In addition, we found for the first time that post-traumatic hypothermia leads to a reduction in the numbers of immunoreactive oligodendrocytes lost following normothermic TBI in specific brain structures. Taken together, these results indicate that post-traumatic hypothermia may improve the acute and more chronic histopathological and functional consequences of TBI by protecting against oligodendrocyte cell death and subsequent demyelination.
Oligodendrocytes are known to play an important role in the various functions of the central nervous system. Experimental and clinical studies have emphasized the detrimental effects of demyelination due to oligodendrocyte cell death in a variety of neurological problems (Fields, 2008). Alterations in the integrity of axonal myelin insulation, for example, lead to functional decline in patients with multiple sclerosis as well as with normal aging (Kujala et al., 1997; Gootjes et al., 2004). The present results are therefore important in that they demonstrate that experimental TBI leads to oligodendrocyte vulnerability that may account for some of the detrimental consequences of TBI that have been documented both experimentally and clinically. Most importantly, therapeutic interventions that protect oligodendrocytes from dying may therefore improve functional outcome in conditions of TBI.
The present study also investigated potential mechanisms for oligodendrocyte vulnerability after CNS injury (McDonald et al., 1996; Crowe et al., 1997; Dewar et al., 2003; Salter and Fern, 2005; Schulz et al., 2008; Domercq et al., 2010; Yu et al., 2009). In previous experimental studies, evidence for apoptotic cell death has been associated with oligodendrocytes in models of TBI (Conti et al., 1998; Shaw et al., 2001; Raghupathi et al., 2002). For example, apoptotic terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive oligodendrocytes were reported by Conti and colleagues several days after experimental TBI (Conti et al., 1998). TUNEL-positive cells have also been identified in both gray and white matter structures from head-injured patients that survived hours to days after injury (Shaw et al., 2001). Bcl-2 staining in a sub-population of apoptotic-appearing TUNEL-positive cells was also reported by Raghupathi et al. (2002) in both gray and white matter structures after TBI. In the current model of moderate FP brain injury, evidence for Caspase 3 immunoreactive cells throughout the traumatized hemisphere has been reported by Keane et al. (2001). In that study, Caspase 3 immunoreactive cells were observed as early as 6 hours after injury with double-labeling approaches showing that some of these cells appeared to be oligodendrocytes. In the current study, double-labeling approaches using cell-specific markers helped demonstrate that oligodendrocyte vulnerability was associated with apoptotic cell processes specifically activated Caspase 3. Subpopulations of oligodendrocytes in both gray and white matter structures at 3 or 7 days after TBI were immunoreactive for this pro-apoptotic marker. In this study we did not utilize quantitative approaches to evaluate the frequency of Caspase 3-positive oligodendrocytes in vulnerable brain regions. In addition to apoptotic cell death, oligodendrocytes may also be dying by other cell death mechanisms (Raghupathi, 2004). Nonetheless, the present quantitative findings do support and extend published TBI data indicating that oligodendrocytes are vulnerable after injury and that a subpopulation may die by apoptotic mechanisms.
Moderate hypothermia has been shown in a variety of experimental models of TBI to promote neuroprotection and improve functional outcome (for review see Dietrich et al., 2009). In 1991, Clifton et al. (1991) reported that moderate hypothermia improved beam walking deficits and reduced mortality after FP injury. In 1994, Dietrich et al. (1994) demonstrated that post-traumatic hypothermia reduced contusion volume and increased the number of surviving neurons within the peri-contusional cerebral cortex. Hypothermia has also been shown to protect the blood–brain barrier from permeability abnormalities (Jiang et al., 1992; Lotocki et al., 2009) as well as reduce the number of damaged axons (Koizumi and Povlishock, 1998; Buki et al., 1999).
In the present study, we report that in addition to these structural benefits, post-traumatic hypothermia also improved the survival of CC1 immunoreactive oligodendrocytes. At both 3 and 7 days after injury, post-traumatic hypothermia was shown to significantly increase the number of immunoreactive oligodendrocytes within the cerebral cortex as well as white matter tracts, including the corpus callosum and external capsule. The preservation of oligodendrocytes in these vulnerable brain regions represents another benefit of moderate hypothermia. It should be stressed that our study was designed only to assess hypothermic protection up to 7 days postinjury. Previous hypothermia investigations have reported that restricted periods of cooling may only transiently protect neurons from dying after an ischemic or traumatic insult (Dietrich et al., 1996). It will therefore be important in future studies to evaluate whether long-term oligodendrocyte survival is seen with this hypothermic protocol or whether longer cooling durations will be necessary to protect these myelin producing cells from irreversible injury.
Hypothermia has previously been shown by Western blotting to reduce Caspase 3 activation after FP brain injury (Lotocki et al., 2006). In the present study, we utilized double-labeling immunocytochemical approaches that identified a subpopulation of Caspase 3 immunoreactive oligodendrocytes after TBI. Interestingly, post-traumatic hypothermia appeared to reduce the overall frequency of oligodendrocytes stained with Caspase 3. Because of the importance of oligodendrocytes in maintaining appropriate myelination of central axons with one oligodendrocyte myelinating many axons, enhanced oligodendrocyte survival could participate in the reported benefits of therapeutic hypothermia on functional recovery under experimental and clinical traumatic conditions (Clifton et al., 1991; Bramlett et al., 1995; Clark et al., 1996; Dixon et al., 1998; Polderman, 2008).
As previously mentioned, significant atrophy of both gray and white matter structures has been reported in this trauma model months to years after injury (for review see Bramlett and Dietrich, 2007). Patterns of progressive atrophy of cortical and subcortical structures along with thinning of white matter tracts and decreased myelin staining has been reported (Bramlett and Dietrich, 2002). In one study, post-traumatic hypothermia reduced the degree of atrophy seen at 2 months after FP brain injury (Bramlett et al., 1997a). Based on the present findings, the benefits of post-traumatic cooling on reducing chronic white matter atrophy may in addition to reducing DAI, also protect oligodendrocytes from cell death and thereby protect myelinated axons in subcortical white matter tracts. It will be important in future studies to clarify whether patterns of demyelination occur as a result of primary oligodendrocyte cell death and/or secondary to Wallerian degeneration, resulting in subsequent axonal pathology and demyelination.
In summary, our findings demonstrate a high degree of oligodendrocyte vulnerability after moderate FP brain injury. These quantitative findings emphasize the potential role of oligodendrocyte cell death in the clinical consequences of TBI. Oligodendrocytes and the maintenance of axonal myelination play a major role in the function of the nervous system, including cognitive development in children (Mabbott et al., 2006). In this regard, diffusion tensor imaging has recently shown the importance of the integrity of white matter in relationships to cognitive ability (Kraus et al., 2007; Kumar et al., 2009; Lo et al., 2009). Thus, there is a potential for abnormalities in myelination after TBI participating in the behavioral deficits seen in patients experiencing higher cognitive dysfunction (Fields, 2008). Our current data report patterns of oligodendrocyte vulnerability and the beneficial effects of post-traumatic hypothermia on the survival of these myelin producing cells. Treatment strategies, including therapeutic hypothermia that target oligodendrocyte vulnerability after TBI, may represent a clinically relevant therapeutic target for future interventions.
Footnotes
Acknowledgments
The authors thank Jeremy Lytle for help and editorial assistance with the article. This work was supported by NS030291 and NS042133.
Disclosure Statement
The authors declare that no competing financial interests exist.