
Abstract
We first assessed the neuroprotective efficacy of mild hypothermia (35°C/24 hours) alone and in combination with magnesium (intravenous loading dose: 360 μmol/kg; 48 hours infusion: 120 μmol/kg/h) commencing 4 hours after global ischemia. Treatment with mild hypothermia alone (CA1 survival rate: 8.7%±0.9%) or with magnesium (9.0%±2.9%) did not significantly increase hippocampal CA1 neuronal survival compared with saline-treated controls (7.1%±0.7%). Next, we assessed mild hypothermia (35°C/24 hours) and moderate hypothermia (33°C/24 hours), alone or in combination with magnesium, when commenced 2 hours after global ischemia. At this time point, all treatments significantly increased CA1 neuronal survival compared with saline controls (CA1 survival rates: mild hypothermia: 13.6%±1.8%; mild hypothermia + magnesium: 19.4%±8.7%; moderate hypothermia: 15.9%±4.1%; moderate hypothermia + magnesium: 21.1%±11.2%; saline control: 6.7%±1.6%; p<0.001). Although a trend for increased neuroprotection was observed when hypothermia was combined with magnesium, it did not reach statistical significance. It also appears that when a 24-hour hypothermia treatment is commenced earlier (≤2 hours postischemia) there is no difference in efficacy between mild and moderate hypothermia.
Introduction
Our laboratory has shown that, using a rat model of global cerebral ischemia, postischemic treatment with milder hypothermia (35°C/24 hours) than that used clinically is also effective at reducing injury to hippocampal CA1 neurons (Zhu et al., 2005). In addition, we have found that the efficacy of mild hypothermia is substantially enhanced when combined with an infusion of magnesium following both global and focal cerebral ischemia (Zhu et al., 2004a, 2004b, 2005; Campbell et al., 2008a, 2008b; Meloni et al., 2009). For its part, magnesium is an ideal treatment to use in combination with hypothermia as it is cheap, has a well-established safety profile, and has a number of potentially beneficial effects on the ischemic brain (Gorelick and Ruland, 2004; Meloni et al., 2006). Magnesium also aids hypothermia induction, improves comfort for awake patients by lowering the shivering threshold (Wadhwa et al., 2005; Meloni et al., 2009), and counteracts the risk of hypothermia-induced hypomagnesaemia (Polderman et al., 2001).
In our present study, our aim was to compare the relative efficacies of mild (35°C) and moderate (33°C) hypothermia with and without magnesium in the two-vessel occlusion with hypotension model of global cerebral ischemia. Although increasing the depth of hypothermia does tend to improve its neuroprotective efficacy, especially when induced during ischemia and for short periods, there are a number of advantages in keeping the depth of hypothermia to a minimum. Mild hypothermia of 35°C has considerable practical advantages over moderate hypothermia of 33°C: it is easier and faster to induce, is achievable in awake patients (Feigin et al., 2002; Meloni et al., 2009), avoids the need for intubation and ventilation, and is associated with less severe side effects (cardiac arrhythmias, hypotension, coagulopathies, and infections). Besides comparing the two levels of hypothermia, another aim was to expand on our previous work and determine a therapeutic time window for mild hypothermia and magnesium combination treatment in our global ischemia model.
Materials and Methods
Global cerebral ischemia model (two-vessel occlusion + hypotension)
This study was approved by the Animal Ethics Committee of the University of Western Australia and conducted according to guidelines established for the use of animals in experimental research as outlined by the Australian National Health and Medical Research Council.
Adult male Sprague-Dawley rats aged 8–10 weeks were fasted overnight, but allowed water ad libitum. Animal surgery was always performed in the morning between 6 and 11 a.m. Anesthesia was induced with 4% isoflurane/27% O2/balanced N2O. The animals were intubated, paralyzed with pancuronium (0.02 mg intravenous [IV] bolus), and ventilated on a rodent ventilator (Ugo Basile). Anesthesia was maintained with 2% isoflurane/27% O2/balanced N2O. For continuous monitoring of arterial blood pressure and blood sampling during ischemia, the tail artery was catheterized with polyethylene tubing (PE-40) primed with heparinized saline (15 U/mL). Via a ventral midline neck incision, the common carotid arteries were exposed and silk thread was placed loosely around the isolated arteries in readiness for subsequent clipping. For induction of hemorrhagic hypotension during ischemia, the right internal jugular vein was cannulated with a length of PVC tubing (PE-50) primed with heparinized saline. Both cranial and rectal temperatures were measured via a thermocouple (Physitemp) and were maintained at 37.5°C±0.2°C before and during ischemia with a heating pad and fan. On recovery after ischemia, core animal body temperature was maintained at 37.5°C±0.2°C using a telemetric temperature control system (see later). A bipolar electroencephalogram (EEG) was recorded using two active lateral subdermal needle electrodes (right and left frontoparietal) and a reference central electrode, which were interfaced with a bioamplifier (AD Instruments). The cutoff frequencies for EEG recordings were set at 0.3 and 10 Hz for the high- and low-pass filters, respectively. EEG and arterial blood pressure were recorded with a PowerLab data acquisition system (AD Instruments). Plasma glucose levels were measured with a blood glucose meter (Miles Laboratories). Ten minutes before the ischemic insult, PaCO2, PaO2, and pH were measured with a pH/blood gas analyzer (ABL5 Radiometer). PaCO2 was maintained between 35 and 45 mmHg, PaO2 between 90 and 120 mmHg, and pH between 7.35 and 7.45 by adjusting ventilation. Anesthetic concentration was reduced to 1% isoflurane/27% O2/balanced N2O just before ischemia. Immediately upon EEG recovery from isoflurane, forebrain ischemia was induced by withdrawing venous blood into a syringe containing 0.5 mL heparinized saline and bilaterally occluding the common carotid arteries using aneurysm clips. A mean arterial blood pressure of 37±2 mmHg was maintained during the ischemia period. The 8-minute ischemic period was recorded from the time when the EEG became isoelectric (with or without some burst activity). After the 8-minute period, the aneurysm clips were removed and the warm (37.5°C) blood was reinfused. EEG recordings were continued postischemia (for a minimum of 15 minutes), and isoflurane was increased to 2% after the reappearance of burst suppression spikes. Blood gases and glucose measurements were then repeated. Arterial and venous lines were removed and the wounds were sutured. On return of spontaneous respiration, isoflurane was discontinued, animals were extubated, allowed to recover, and returned to their cages with free access to food and water. Cages were housed in a temperature-controlled holding box (see below for details) for 26–28 hours, after which time they were transferred to a holding room maintained at 27°C. Sham-operated animals received identical anesthesia, surgery, and holding environments as experimental animals but were not rendered ischemic. Animals were weighed daily until sacrifice.
Experimental groups
Two exclusion criteria were established based upon previous experience with this model under anesthesia with isoflurane (Li et al., 2011): (i) all rats for which the isoelectric EEG time after ischemia (the time from the beginning of reperfusion until the return of spontaneous spike activity) was no longer than 12 minutes were excluded; and (ii) rats for which the isoelectric EEG time after ischemia was between 12 and 14 minutes and for which EEGs during ischemia exhibited intermittent burst activity were excluded. In all of the studies, rats were assigned to treatment groups by random number tables, and the operators were blind to treatment (except hypothermia/normothermia) until all data were collected.
Trial 1 consisted of three groups: (i) IV saline (0.9% NaCl) and controlled normothermia (37.5°C±0.2°C); (ii) IV saline and mild hypothermia (35.0°C±0.2°C); and (iii) IV magnesium (MgSO4·7H2O 360 μmol/kg loading dose; 120 μmol/kg/h infusion) and mild hypothermia. All treatments were commenced 4 hours after ischemia. During the 4-hour interval after ischemia and before treatment commencement, animal body temperature was actively maintained at 37.5°C±0.2°C. Temperature control (normothermia and hypothermia) was continued for 24 hours, whereas IV infusions were continued for 48 hours.
Trial 2 consisted of five groups: (i) IV saline and controlled normothermia (37.5°C±0.2°C); (ii) IV saline and mild hypothermia (35.0°C±0.2°C); (iii) IV magnesium and mild hypothermia; (iv) IV saline and moderate hypothermia (33.0°C±0.2°C); and (v) IV magnesium and moderate hypothermia. All treatments were commenced 2 hours after ischemia. During the 2-hour interval after ischemia and before treatment commencement, animal body temperature was actively maintained at 37.5°C±0.2°C. Temperature control was continued for 24 hours, whereas IV infusions were continued for 48 hours.
Intravenous infusions and postischemic maintenance of animal body temperature
Intravenous infusions were administered via the right internal jugular vein using an animal tethering system (Instech Laboratories) as previously described (Miles et al., 2001; Zhu et al., 2004a, 2004b).
Animal body temperatures were measured and controlled using a telemetric recording system, consisting of an implantable radio-transmitting temperature probe and computer data acquisition as previously described (Zhu et al., 2005). Briefly, thermistor probes were inserted into the abdominal cavities of the animals at the time of surgery to induce global ischemia, which remain in place until animals were sacrificed. Probes had previously been calibrated in a 37°C incubator, while in a beaker of water and placed next to a commercial temperature probe (Physitemp) to ensure correct temperature settings. During the temperature control period, animals were placed in a cage (containing food and water) housed within an insulated box in a 4°C cold room. Animal body temperatures were regulated by a heating/cooling fan and, when necessary, a water mister. Rewarming to 37°C occurred over a 60-minute period. An overview of the timeline of ischemia and treatment interventions is shown in Figure 1.

Timeline of experimental procedures and treatment interventions following global cerebral ischemia (not to scale).
Histological analysis
At 7 days postischemia, rats were anesthetized with pentobarbital (100 mg/kg IP) and transcardially perfused with 200 mL of normal saline followed by 200 mL of 4% neutral buffered formalin. The brains were removed and postfixed for 1 week before being paraffin embedded. The brains were sectioned at 5 μm thickness at bregma section −3.8 mm according to a standard rat brain atlas (Paxinos and Watson, 1998) and stained with cresyl violet. The number of normal-appearing pyramidal neurons per high-power field (400×) in 1000 μm segments in the medial, intermediate, and lateral sections of the left and right hippocampal CA1 regions were counted giving a representative CA1 neuronal count for each animal (Zhu et al., 2004b, 2005). CA1 neuronal survival rates were expressed as a percentage of neuronal cell counts in sham animals, which was taken as 100%. Neuronal cell counts were performed by an observer who was blinded to the experimental groups.
Statistics
Histological data analysis was by analysis of variance, followed by post hoc Bonferroni/Dunn. The data from Trial 2 violated the assumption of equal variance and were therefore transformed logarithmically before statistical analysis to achieve normal distributions. Statistical significance was assumed at p<0.05.
Results
Physiological data
Other than mean arterial blood pressure, which was controlled at 37±2 mmHg during the 8 minutes of ischemia, all other physiological variables remained normal and there were no statistically significant differences between experimental groups for Trials 1 and 2 (Tables 1 and 2).
Data are given as mean±SD.
PaO2, arterial blood oxygen partial pressure; PaCO2, arterial blood carbon dioxide partial pressure; MABP, mean arterial blood pressure; SD, standard deviation.
Data are given as mean±SD.
Postischemic temperature control
In Trial 1, animal body temperatures were decreased to 35.0°C±0.2°C over 20 minutes, commencing 4 hours after ischemia, and were maintained at this temperature for 24 hours in hypothermia-treated animals. In Trial 2, core body temperatures were reduced to either 35.0°C±0.2°C or 33.0°C±0.2°C over 20 minutes, commencing 2 hours after ischemia, and maintained for 24 hours in hypothermia-treated animals. Representative graphs showing body temperature over different periods of mild or moderate hypothermia are provided in Figure 2A–C.
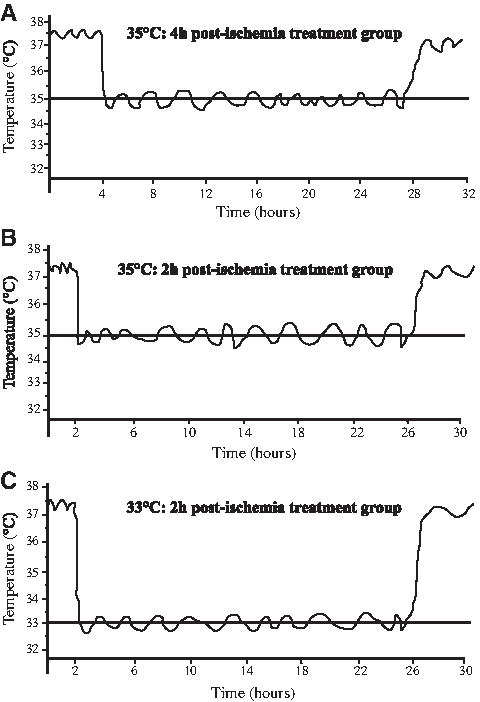
Representative graphs of core body temperature measurements from individual rats treated with mild or moderate hypothermia.
CA1 neuronal survival in treatment groups
Trial 1
In animals treated with mild hypothermia alone or in combination with magnesium starting 4 hours postischemia, CA1 neuronal survival rates were 8.7%±0.9% (mean±standard deviation) and 9.0%±2.9%, respectively. These neuronal survival rates did not differ significantly from saline-treated controls (7.1%±0.7%; Figs. 3 and 4A).
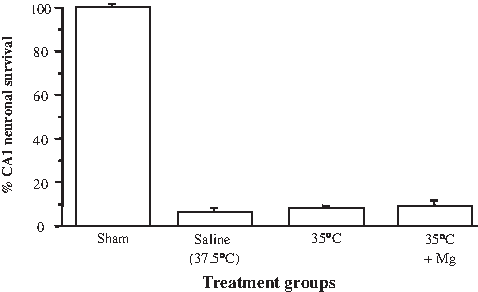
Effects of mild hypothermia and magnesium administered alone or in combination on CA1 neuronal survival following global ischemia assessed at 7 days (Trial 1). Mild hypothermia and magnesium administrated 4 hours postischemia. Values are the mean (±standard deviation [SD]) numbers of normal neurons counted in segments of the CA1 region of the hippocampus.
Photomicrographs of cresyl violet-stained sections of CA1 hippocampal neurons from a nonischemic rat and rats subjected to global ischemia assessed at 7 days.
Trial 2
In animals treated with mild or moderate hypothermia starting 2 hours postischemia, CA1 neuronal survival rates were 13.6%±1.8% and 15.9%±4.1%, respectively, both significantly higher than the 6.7%±1.6% CA1 neuronal survival in saline-treated controls (p<0.05; Figs. 4B and 5). In animals treated with mild or moderate hypothermia and magnesium, CA1 neuronal survival rates were 19.4%±8.7% and 21.1%±11.2%, respectively, which were also significantly higher than controls (p<0.05; Figs. 4B and 5). The level of CA1 neuronal survival did not differ significantly between other treatment groups.
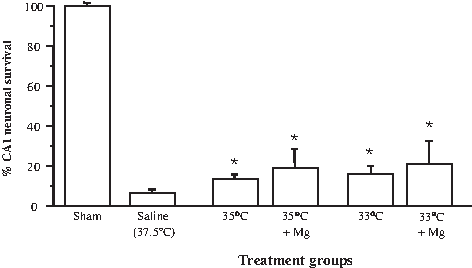
Effects of mild hypothermia and magnesium administered alone or in combination on CA1 neuronal survival following global ischemia assessed at 7 days (Trial 2). Mild or moderate hypothermia with and without magnesium administrated 2 hours postischemia. *p<0.05 compared with normothermic saline controls. Values are the mean (±SD) numbers of normal neurons counted in segments of the CA1 region of the hippocampus.
Animal mortality
Trial 1
Mortalities in each group were as follows: mild hypothermia 1 (on day 1 postischemia), mild hypothermia/magnesium 0, and saline/normothermia 1 (on day 2).
Trial 2
Mortalities in each group were as follows: moderate hypothermia/magnesium 1 (on day 6), mild hypothermia/magnesium 1 (on day 3), mild hypothermia 1 (on day 5), moderate hypothermia 0, and saline/normothermia 1 (on day 1).
Discussion
We have previously shown that magnesium treatment and 24 hours of mild hypothermia, induced by 2 hours after global ischemia (i.e., with a 20 minutes cooling period starting at 1 hour 40 minutes after ischemia), increases CA1 neuronal survival from 5% to 76%; under the same conditions, hypothermia alone increases neuronal survival from 5% to 43% (Zhu et al., 2005). In a focal ischemia model, combined magnesium and mild hypothermia reduced infarct volumes by 39% when commenced as late as 4 hours after permanent middle cerebral artery occlusion (Campbell et al., 2008a). Thus, in Trial 1 our aim was to use the global ischemia model to investigate neuroprotection at the 4 hours time point, comparing mild hypothermia/magnesium with mild hypothermia alone. However, we found no evidence of neuroprotection at this time point for either mild hypothermia/magnesium or mild hypothermia treatments. We repeated a small-scale trial with treatment commencing 3 hours after ischemia, and again no neuroprotection was observed for either group (data not shown).
Trial 2 investigated mild hypothermia/magnesium and mild hypothermia treatment starting at 2 hours after ischemia (i.e., with a 20-minute cooling period starting at 2 hours after ischemia). Although there was a statistically significant treatment effect, the level of neuroprotection from mild hypothermia was smaller (14% vs. 43%) than previously reported, and there was no significant synergistic benefit from magnesium (Zhu et al., 2005). We believe differences in model conditions and the timing of hypothermia induction have contributed significantly to these different findings. In our present study, the following model conditions applied: (1) isoflurane was used instead of halothane (as halothane has become unavailable in Australia); (2) during cerebral ischemia, the target blood pressure was 37 mmHg instead of 45 mmHg; (3) during ischemia, temporal muscle and rectal temperatures were kept at 37.5°C±0.2°C instead of 37.0°C±0.2°C; and (4) during the interval after ischemia and treatment with mild hypothermia and/or magnesium, animal body temperature was actively maintained at 37.5°C±0.2°C with the temperature control system, instead of allowing self-regulation at 37.0°C±0.5°C with minimal intervention (warm environment and external heating if necessary). To this end, it is possible that we may have inadvertently overheated the brain by 0.5°C–1°C above our target temperature, as a previous study has shown a disproportionate temperature difference between core and brain during heating (Plahta et al., 2004). In addition, in Trial 2 of the present study, we induced hypothermia commencing 2 hours after ischemia, whereas in our previous study hypothermia had been already achieved by 2 hours. Thus, it is not unreasonable to conclude that the present model is associated with more severe ischemic and postischemic conditions, and combined with the longer delay for hypothermia induction, the efficacy of mild hypothermia and the time window in which magnesium exerts a synergistic effect is reduced.
To examine this hypothesis, we have shown that under current model conditions (isoflurane, blood pressure 37 mmHg), if cranial temperature is maintained at 37.0°C±0.2°C during ischemia, core body temperature at 37.0°C±0.2°C postischemia, and both mild hypothermia target temperature and commencement of magnesium treatment are achieved by 2 hours postischemia, the level of CA1 survival increases to 55% (Li et al., 2011). Hence, these findings demonstrate how small changes in model conditions and treatment parameters can have a dramatic influence in terms of neuroprotective outcome.
Trial 2 also investigated whether moderate hypothermia was more efficacious than mild hypothermia starting 2 hours after ischemia. By the measure of neuronal survival in the hippocampal CA1 region, there was no difference between the treatments. In addition, although there appeared to be a trend for increased neuroprotection when mild or moderate hypothermia was combined with magnesium, it did not reach statistical significance. These findings are significant, because, to induce and maintain moderate hypothermia (33°C) in clinical patients, cooling equipment has to be used in conjunction with sedatives, muscle relaxants, and ventilators in an intensive care unit (Schwab et al., 1998; van der Worp et al., 2010). Mild hypothermic (35°C) treatment, by comparison, is easier to achieve, and maintenance requires less resources and is more amenable in awake patients (Kammersgaard et al., 2000; Meloni et al., 2009). In addition, hypothermia-associated side-effects such as impairment of immune responses and predisposition to coagulopathies would be less (Logue et al., 2007). Thus, based on our findings and the advantages of mild hypothermia over moderate hypothermia, it would be of significant benefit if future hypothermia trials were designed to directly compare the two levels of hypothermia.
Another factor that needs to be considered is whether the CA1 neuronal protection observed in the present study is long lasting. This is an important issue as previous studies have shown that, in some cases, treatments including hypothermia merely delay rather than prevent CA1 neuronal loss (Dietrich et al., 1993; Shuaib et al., 1995). In addition, it would be of interest to determine whether there is a differential effect of mild versus moderate hypothermia in preserving long-term neuronal survival.
As mentioned earlier, although the global ischemia model used in the present studies was similar to the one used in our earlier studies (Zhu et al., 2005), there were some differences. The change of anesthetic agent to isoflurane brought with it some unanticipated problems, which are discussed at length in a recent paper (Li et al., 2011). Briefly, when using halothane, 8 minutes of cerebral ischemia with hypotension of 45 mmHg produced a consistent CA1 hippocampal injury such that neuronal survival was between 3% and 7%. However, when isoflurane was used, the same ischemic period and blood pressure produced a highly variable lesion. By changing blood pressure to 37 mmHg and by incorporating analysis of intra- and postischemic EEG, the consistency of the model was greatly improved and the neuronal survival rates (5%–9%) were highly comparable than in earlier experiments. Although it is difficult, if not impossible, to make definitive statements as to what is a normal body temperature (varies with site and method of measurement, degree of stimulation, and time of day), by many estimates the normal core temperature for a rat is closer to 37.5°C than 37.0°C. In recognition of this, we used 37.5°C as the target temperature during ischemia and in the period between ischemia and induction of hypothermia. Although the difference is small, minor changes in brain temperature during and after ischemia can have profound effects on ischemic outcome (Busto et al., 1987; Wass et al., 1995; Plahta et al., 2004). We note that in a recent global ischemia study by Silasi and Colbourne (2011), rats were maintained at ≈36°C during a 1, 4, 12, or 24-hour delay before hypothermia therapy (33°C/24 hours +35°C/24 hours +36°C/24 hours), which resulted in CA1 neuronal protection levels ranging from 50% to 95%. It should also be mentioned that in a clinical situation there would be little benefit in actively maintaining patients normothermic before hypothermia therapy.
In conclusion, our results demonstrate that brain protection from combined hypothermia and magnesium treatment has a clear, and short, time window under our current experimental global cerebral ischemia model conditions. It should be mentioned, however, that other studies have demonstrated a wider therapeutic window and higher efficacy with prolonged durations (48–72 hours) of moderate and/or mild hypothermia (Colbourne et al., 1998, 1999a, 1999b; Silasi and Colbourne, 2011). Further, we have revealed that mild hypothermia (35°C/24 hours) had the same protective effect on neuronal survival as that of moderate hypothermia (33°C/24 hours), but it resulted in less body weight loss compared with that of moderate hypothermia when magnesium treatment was given. In addition, we have highlighted how minor changes in cerebral ischemia model parameters and timing of treatment can have a considerable effect on neuroprotective outcome.
Footnotes
Acknowledgments
The authors acknowledge Joanne Chieng and Russell Johnsen for technical assistance. This study was supported by the Neuromuscular Foundation of Western Australia and by a grant from the National Health and Medical Research Council of Australia.
Disclosure Statement
The authors declare that no competing financial interests exist.