
Abstract
Hypothermia is an established form of treatment employed following cardiac arrest to limit cerebral injury. The question addressed in this review is whether it is possible to use hypothermia to protect the heart during ischemia resulting from ST-elevation myocardial infarction (STEMI). Mild hypothermia (32°C–35°C) may be of benefit as an adjunctive treatment for STEMI by reducing the extent of the infarct and the effects of the four components of ischemia reperfusion injury: myocardial stunning, microvascular obstruction, reperfusion arrhythmia, and lethal reperfusion injury. To reduce cerebral injury after cardiac arrest, hypothermia can be initiated after reperfusion, and should be maintained for 24–48 hours. However, evidence suggests that to protect the heart in cases of STEMI, hypothermia should be initiated as early as possible after the onset of ischemia, at least before reperfusion. Clinical and experimental results indicate that it is of paramount importance to achieve a body temperature below 35°C before reperfusion to reduce the size of the infarct in the treatment of STEMI patients, and that treatment need only be continued for a relatively short period after reperfusion. Hypothermia has wide-ranging effects on most of the mechanisms involved in ischemia and reperfusion injury, which may explain the potent, highly reproducible cardioprotective effects seen in a large number of studies in different species. Subjecting conscious patients with STEMI to hypothermia is safe, feasible, and well tolerated, but antishivering strategies must be employed. Clinical studies are ongoing to evaluate hypothermia as an adjunctive treatment for myocardial infarction. This review discusses the experimental basis for using mild hypothermia to provide cardioprotection, methods of inducing hypothermia, the timing and duration of treatment, and ways in which the knowledge gained can be translated into clinical treatment.
Introduction
Hypothermia is a well-established method of protecting the heart during coronary artery bypass grafting (Varon and Acosta, 2008). It is also crucial for successful heart transplantation as it protects the donor heart after explantation. However, deep hypothermia is often used (<30°C), which may cause spontaneous ventricular fibrillation and reduced cardiac function, leading to the need for circulatory support. To treat conscious patients with STEMI, cardiologists are restricted to the use of mild hypothermia (32°C–35°C). Interestingly, even mild hypothermia has the ability to protect the heart against the development of myocardial infarction. In a large number of animal studies, the protective effect has been found to range from 18% to 90% when hypothermia was started during ischemia, in the rat, rabbit, dog, and pig (Hale and Kloner, 2011; Tissier et al., 2011a). Further, small clinical safety and feasibility studies have shown promising effects (Gotberg et al., 2008). This review discusses the experimental basis for using mild hypothermia to provide cardioprotection, methods of inducing hypothermia, the timing and duration of treatment, and ways in which the knowledge gained can be translated into clinical treatment.
Cardioprotection—the Next Treatment Opportunity
Reperfusion injuries and cardioprotection
The current treatment of acute STEMI is to open the occluded coronary artery and to reperfuse the ischemic myocardium using either thrombolysis or primary percutaneous coronary intervention (PCI) as soon as possible, to reduce the extent of the infarction and associated complications. Infarct size is one of the main predictors of both short- and long-term outcome in patients with acute myocardial infarction (AMI) (Miller et al., 1995; Burns et al., 2002). Additional antithrombotic therapy can be used to help keep the artery open. Paradoxically, the actual process of restoring coronary blood flow to previously ischemic myocardium can in itself increase the myocardial injury sustained during ischemia—a phenomenon termed myocardial reperfusion injury (Kloner et al., 1974; Kloner, 1993; Ambrosio and Tritto, 1997). Reperfusion injury causes four types of cardiac dysfunction: (1) myocardial stunning, (2) microvascular obstruction or no-reflow phenomenon, (3) reperfusion arrhythmia, and (4) lethal reperfusion injury (Yellon and Hausenloy, 2007). Lethal reperfusion injury is defined as cardiomyocyte death mediated by reperfusion and not by the ischemia alone. As will be discussed later, hypothermia may have beneficial effects on all these kinds of reperfusion injuries, but the focus of this review is on lethal reperfusion injury.
Promising or failed cardioprotective strategies
Previous attempts to limit this form of myocardial injury in patients after STEMI using pharmacological strategies as adjuncts to primary PCI have often worked well in animal studies and appeared promising in small clinical phase I or II studies, but have generally failed in larger randomized clinical trials. These include treatment with antioxidants (superoxide dismutase, trimetazidine, allopurinol, and edaravone), calcium-channel antagonists (diltiazem and MCC-135), sodium–hydrogen exchange inhibitors (cariporide and eniporide), antiinflammatory agents (anti-CD18, anti-CD11, P-selectin antagonists, and FX06; an inhibitor of vascular endothelial-cadherin and adenosine), complement inhibitors (pexelizumab), protein kinase C-delta-inhibitors (delcasertib), metabolic modulation (glucose, insulin, and potassium), magnesium, nicorandil, atorvastatin, and erythropoietin (Yellon and Hausenloy, 2007; Hausenloy and Yellon, 2011). Some pharmaceutical agents, such as cyclosporine and atrial natriuretic peptide, still appear to be promising according to the results of medium-sized clinical trials (Hausenloy and Yellon, 2011).
A more mechanical approach has been studied, involving conditioning therapies that consist of inducing short ischemic periods that trigger protective responses that are still not fully understood. Preconditioning is highly effective, but is not clinically feasible. Remote preconditioning, in which the circulation to the arm is repeatedly occluded in STEMI patients in the ambulance on the way to the PCI lab, was recently shown to reduce infarct size in a trial including 142 patients (Botker et al., 2010). Postconditioning is a technique in which occlusion is repeatedly performed with the angioplasty balloon directly after opening the vessel. This strategy has been tested in four clinical trials in which infarct size was examined. In a study on 38 patients, the size of the infarct was reduced by 39% when evaluated with single-photon emission comuted tomography (SPECT) after 6 months (Staat et al., 2005; Thibault et al., 2008). Yang and coworkers (2007) found a 27% reduction after 1 week, also using SPECT (n=41). Lonborg and coworkers (2010) evaluated infarct size with cardiac magnetic resonance imaging (MRI) and found a 19% reduction in infarct size after 3 months as a result of postconditioning (n=86). They also found a reduced incidence of heart failure. On the other hand, in one of the larger studies (n=76), in which MRI was also used for infarct size evaluation, no difference was seen in infarct size or troponin levels (Sorensson et al., 2009). Further, a recent study with 79 patients found no significant differences in infarct size and left ventricular (LV) ejection fraction at 1 week and 6 months after MI (Freixa et al., 2011). Neither of these methods can therefore be considered to provide sufficient evidence for general clinical implementation.
As will be discussed later, many mechanisms are involved in reperfusion injury. The problem with previous cardioprotective agents could be that they only affect one single mechanism per treatment. Hypothermia has the advantage of affecting a wide range of mechanisms, and this may be important in achieving clinical effects (Tables 1, 2).
t-PA, tissue-plasminogen-activating factor.
Early Animal Experiments Using Mild Hypothermia for AMI
It has been known for over 30 years that deep hypothermia (<30°C) reduces the extent of a myocardial infarct (Abendschein et al., 1978); however, the detrimental physiological effects on the heart (spontaneous ventricular fibrillation and reduced ventricular function) makes it less suitable for treating conscious patients. The first study to demonstrate cardioprotective effects of mild hypothermia in myocardial ischemia was published in 1994 by Chien and coworkers (Chien et al., 1994). They found a linear correlation between infarct size, in relation to the area at risk, and temperature in the interval from 35°C to 42°C in rabbits subjected to 30 minutes coronary occlusion; the size of the infarct being decreased by 8% for each degree of temperature reduction. Importantly, they investigated whether the effect of hypothermia was mediated by the bradycardia induced by hypothermia (Table 1). To compensate for bradycardia, they paced one group of animals, but the reduction in infarct size was similar in both groups, suggesting that bradycardia was not the mechanism by which hypothermia reduced infarct size.
In a study using an open-chest pig model, the animals were cooled before inducing ischemia (Duncker et al., 1996). Hypothermia was maintained during the whole period of ischemia and for 4 hours during reperfusion. A relationship was also found between temperature and infarct size: increasing the temperature to 39°C increased the size of the infarction, and decreasing it reduced the infarct size. At a temperature of 35°C, complete protection was achieved and no infarction was seen. A similar study in dogs demonstrated a reduction in infarct size from 32% to 17% in the group treated at 35°C–38°C compared with the group treated at 38°C–42°C (Schwartz et al., 1997). Although these preischemia cooling studies are important in demonstrating the powerful cardioprotective effects of hypothermia, the protocol cannot be applied to humans, as patients do not present at the hospital before STEMI occurs. It is important to note that not only does lowering the body temperature below 37°C reduce infarct size, but also elevated temperature increases infarct size, indicating that fever should be treated in patients with AMI (Chien et al., 1994; Duncker et al., 1996; Hale and Kloner, 2002).
Hale and Kloner performed a series of open-chest rabbit experiments in which they started topical cooling during ischemia (Hale et al., 1997; Hale and Kloner, 1997, 1998). Their results clearly showed that the application of hypothermia after the onset of ischemia could substantially reduce infarct size and that the longer the period of hypothermia during ischemia, the better the cardioprotective effect. Several subsequent studies have confirmed the relation between duration of cooling during the ischemic period and outcome (Miki et al., 1998; Tissier et al., 2007, 2009). The combined results of these studies demonstrate an excellent correlation between the duration of cooling during ischemia and infarct size (Tissier et al., 2011a, 2011b). In a review by Tissier and coworkers, the results of 16 studies on the effect of hypothermia during ischemia are summarized in a structured table. A reduction in myocardial infarct size was found in all these studies, ranging from 18% to 90% (Tissier et al., 2011a). These results are important as they demonstrate that inducing hypothermia after the onset of ischemia reduces damage to the heart, and thus that clinically applicable protocols can be developed.
Another aspect of reperfusion injury, myocardial stunning, is also prevented by hypothermia, as has been shown in a rabbit model in which myocardial function, measured as segment length shortening, was significantly increased in the hypothermia-treated animals (Tissier et al., 2009).
A multitude of cooling methods have been used in animal experiments: topical cooling using ice in the open chest (Hale and Kloner, 1997), cold perfusion of the pericardium (Dave et al., 1998), intracoronary infusions (Otake et al., 2007), hypothermic coronary retroperfusion (Wakida et al., 1991), and extracorporeal circuit cooling (Maeng et al., 2006), but most of them are too complex or too dangerous to be used in a conscious patient with myocardial infarction.
A pivotal animal study in the development of hypothermia treatment in the clinical setting was carried out by Dae and coworkers. They used an endovascular heat-exchange cooling catheter designed for clinical use, inserted into the vena cava via the femoral vein, in human-sized pigs subjected to a 60-minute occlusion of the left anterior descending artery (Dae et al., 2002). Cooling was initiated after 20 minutes of ischemia, and resulted in an 80% reduction in the size of the infarct in relation to the area at risk (Dae et al., 2002). The transferability of the protocol and the impressive effect led to the initiation of two large clinical trials, the COOL-MI and the ICE-IT trials.
Early Clinical Trials of Hypothermia for STEMI
Small clinical trials have demonstrated the safety and feasibility of cooling conscious patients with AMI (Dixon et al., 2002; Kandzari et al., 2004). Dixon et al. randomized 42 patients to endovascular heat-exchange cooling or standard PCI treatment (Dixon et al., 2002). Cooling was well tolerated, with no hemodynamic instability or increase in arrhythmia, and a nonsignificant trend toward reduction in infarct size was seen. In the LOWTEMP study, Kandzari et al. treated 18 nonrandomized patients with endovascular cooling as adjunctive therapy to primary PCI (Kandzari et al., 2004). Periprocedural endovascular cooling successfully decreased the core body temperature and was well tolerated. In the NICAMI study, surface cooling was tested in cases of AMI (Ly et al., 2005). Cooling was well tolerated and safe, but it took 79 minutes on average to reach the target temperature showing that surface cooling is too slow for STEMI treatment.
Two clinical trials investigating mild hypothermia using endovascular cooling catheters as an adjunct therapy for STEMI failed to show a reduction in infarct size (Grines, 2004; O'Neill, 2004; O'Neill et al., 2005). In the COOL-MI trial (conducted by Radiant, now Zoll), 392 STEMI patients were randomized to standard PCI or endovascular cooling for 3 hours followed by 4 hours of rewarming, in addition to PCI.9,34 The door-to-balloon time was 18 minutes longer in the hypothermia group. Nearly all the patients subjected to hypothermia (94%) tolerated the complete cooling protocol well; however, no difference was found in the clinical endpoints, serious side effects, or in the primary endpoint; infarct size was measured with SPECT after 30 days (PCI: 13.8% vs. PCI+hypothermia: 14.1%). In a post hoc analysis of anterior infarcts in patients who had reached a temperature below 35°C at the onset of reperfusion, a trend toward a 49% reduction in infarct size was seen (PCI: 18.2%, n=58 vs. PCI+hypothermia: 9.3%, n=16).
In the ICE-IT trial (conducted by InnerCool, now Philips), 228 STEMI patients were randomized to standard PCI or endovascular cooling for 6 hours followed by 3 hours of rewarming, in addition to PCI (Grines, 2004). No significant differences were found in clinical endpoints or serious side effects. A higher number of deaths in the hypothermia group (9 vs. 4) could be explained by an excess of elderly patients in the hypothermia group. However, this affected the primary endpoint infarct size, measured with SPECT after 30 days; as according to the protocol, these patients were imputed to have the largest infarct size. However, when the data were analyzed without imputed values, there was a trend toward a 23% reduction in infarct size in the hypothermia group (PCI: 13.2% vs. PCI+hypothermia: 10.2%, p=0.14). In a post hoc analysis of anterior infarcts in patients who reached a temperature below <35°C at the onset of reperfusion, there was a trend toward a 43% reduction in infarct size (PCI: 22.7%, n=38 vs. PCI+hypothermia: 12.9%, n=10, p=0.09).
Interest in hypothermia as a form of treatment for STEMI was considerably reduced after these trials were presented in 2002 and 2003. The drawback of the studies was that the cooling rate was relatively slow, and only a minority of the patients (∼30%) had reached a temperature below 35°C at the onset of reperfusion. The post hoc analysis of the two trials showing strong trends toward a positive effect in patients who did reach a temperature <35°C was highly interesting, but needed to be verified in animal experiments and prospective clinical trials.
Timing, Speed of Induction, and Duration of Hypothermia
Is cooling before reperfusion the key to success?
As discussed previously, there is ample evidence that inducing hypothermia during the period of ischemia reduces infarct size. However, it is also known that reperfusion injury may account for as much as 50% of the final size of the infarct (Yellon and Hausenloy, 2007). The post hoc analysis of the data from the COOL-MI and ICE-IT trials suggests that hypothermia was only effective if a sufficiently low temperature was achieved before reperfusion. To test this, we first needed a more rapid method of cooling. At that time we already used to cool our cardiac arrest patients with liters of cold saline in the emergency room and on the way to intensive care.
We therefore tried to combine the most rapid cooling method, endovascular cooling, with a fast infusion of 1 L cold saline in 40–45 kg pigs, and were able to cool the pigs to below 35°C within 5 minutes (Gotberg et al., 2008). When cooling was instigated immediately after reperfusion, no effect on infarct size was seen, despite reaching a body temperature of 35°C 5 minutes after reperfusion, confirming the post hoc analysis of the clinical studies regarding the lack of effect of postreperfusion cooling. When hypothermia was initiated 15 minutes before reperfusion during a 40 minutes period of ischemia, the size of the infarct was reduced by 39%. However, this meant that the pigs had received therapeutic hypothermia for 10 minutes of the total 40-minute period of ischemia, and this could have been responsible for the whole effect. To ascertain whether hypothermia has an effect on reperfusion injury per se, the ischemic period was extended by 5 minutes and the extra time was used to induce hypothermia (Gotberg et al., 2011). Thus, the two groups had undergone the same period of normothermic ischemia. The size of the infarct was reduced by 18%, proving that hypothermia has a separate effect on reperfusion injury, independent of the effect on ischemia (Gotberg et al., 2011). This 18% reduction is probably an underestimation as the comparison was made against a 5-minute shorter period of ischemia in the control group. In this way, we confirmed the results of the post hoc analysis of the data from the COOL-MI and ICE-IT studies that hypothermia is effective before reperfusion and is without effect after reperfusion.
The effects of hypothermia immediately before and after reperfusion have been examined in other animal studies, but the cooling methods were often too slow to achieve therapeutic temperatures before reperfusion (Maeng et al., 2006), or the statistical significance was only borderline (Otake et al., 2007). Tissier and coworkers applied rapid liquid ventilation cooling in a rabbit model just before reperfusion and succeeded in reaching therapeutic temperatures without observing any effect on infarct size (Tissier et al., 2007, 2011b). In contrast, a reduction in infarct size was found in another rabbit study when hypothermia was initiated 5 minutes before reperfusion (Kanemoto et al., 2009). There could be several reasons for these conflicting findings. Since the pig model is more similar to man than the rabbit model, it is more clinically relevant, and the positive effects on reperfusion injury seen in the pig model will hopefully also be found in patients.
Duration of hypothermia
The evidence shows that cooling should be achieved as early as possible during ischemia, and should reach the minimum before reperfusion. However, it is still not known how long the cooling should continue after reperfusion. This question also remains unanswered in the case of hypothermia for cardiac arrest. We extended the postreperfusion cooling in our pig model from 15 to 60 minutes postreperfusion, without finding any additional protecting effects on infarct size (Gotberg et al., 2011). The finding is in agreement with the lack of effect of hypothermia when instigated after the start of reperfusion. This indicates that hypothermia could be terminated quite early after reperfusion. We concluded that the 3–6 hours of postreperfusion cooling used in the COOL-MI and ICE-IT studies is unnecessarily long and could be shortened to 1 hour. At the same time, it is important that the subject's whole body is thoroughly cooled. Cold saline alone cools the blood quickly, but gives a rapid rebound in temperature resulting in a lack of effect on infarct size (Gotberg et al., 2011), demonstrating that a combination with endovascular cooling is necessary.
Microvascular Obstruction
Every coronary interventionist performing primary PCI is aware of, and often frustrated by, a phenomenon called no-reflow or TIMI 2 flow. Upon opening the occluded epicardial coronary artery, a good blood flow is obtained (TIMI 3). However, within a few minutes, the flow is restricted by poor runoff in the microcirculation. Histologically, this represents a microvascular obstruction (Jaffe et al., 2008). The mechanism is not fully understood, but involves inflammatory and complement activation, endothelial swelling, red blood cell extravasation, and tissue edema. The phenomenon is prevalent in large myocardial infarctions and is correlated to the duration of the myocardial infarction (Lima et al., 1995).
The presence of microvascular obstruction is independently associated with impaired recovery of LV function and a poor long-term clinical outcome (Wu et al., 1998; Choi et al., 2004). Hale et al. found that hypothermia reduced microvascular obstruction (measured with microspheres) to a greater extent than the reduction in infarct size in rabbits (Hale et al., 2003; Gotberg et al., 2008). This phenomenon was confirmed in our pig model, where hypothermia reduced the infarct size by 39%, while microvascular obstruction was completely abolished (Gotberg et al., 2008). Further, hypothermia initiated postreperfusion or by cold saline alone reduced microvascular obstruction although it did not result in any reduction in infarct size (Gotberg et al., 2008, 2011). These findings indicate that infarct size and microvascular obstruction are, at least partly, separate mechanisms and link microvascular obstruction to reperfusion injury (Fig. 1).
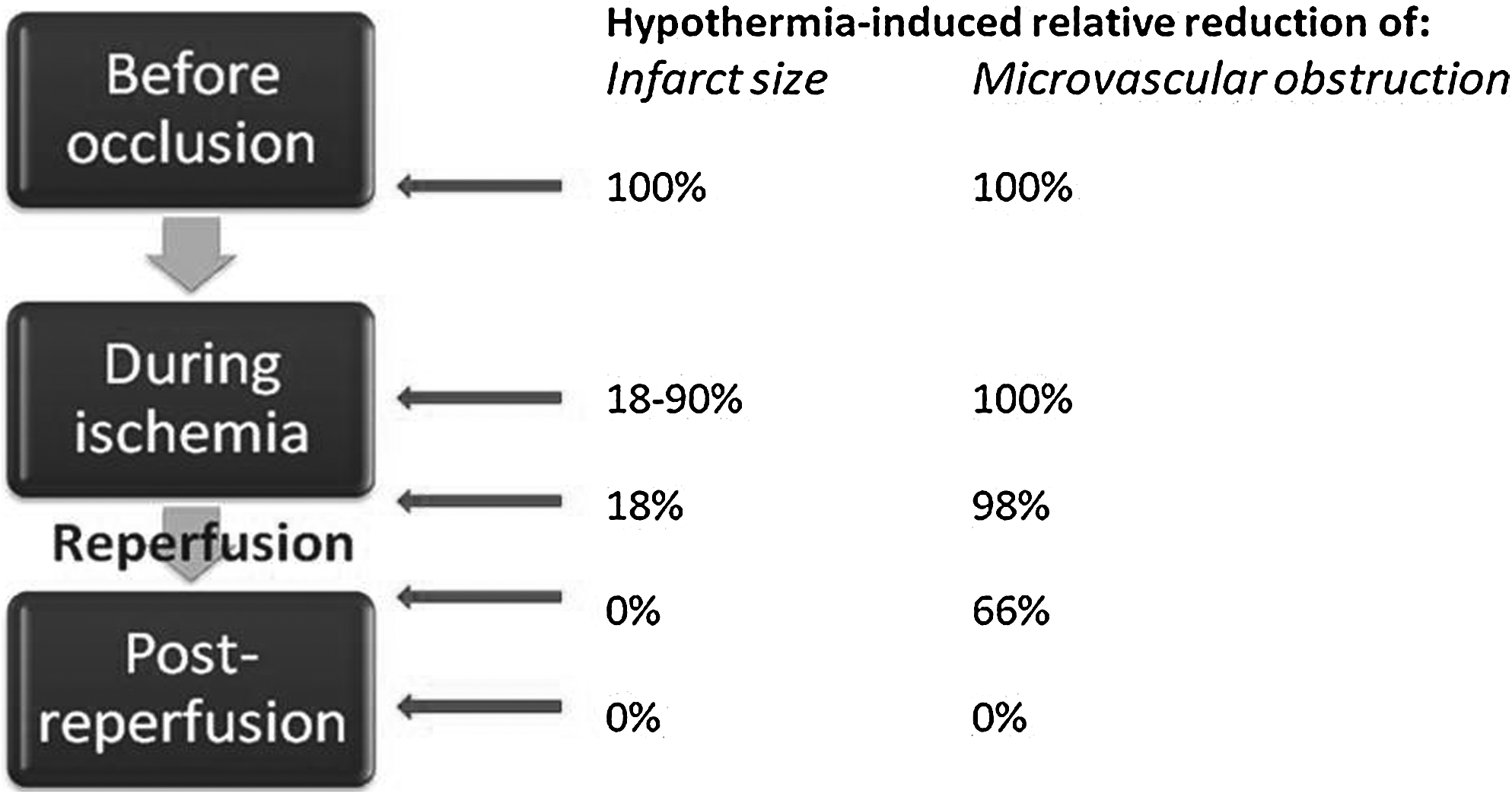
Timing of hypothermia. Relative reduction in infarct size and microvascular obstruction by hypothermia depends on when hypothermia is achieved. The values are estimations based on different animal experiments (see text for references).
Infarct Morphology
The high resolution of ex vivo cardiac MRI of pig hearts revealed a “speckled” infarct morphology with scattered myocardial salvage in the hypothermia-treated group (Gotberg et al., 2008). This is in agreement with the findings of Dae and coworkers (2002), who described scattered islands of reduced Sestamibi uptake in pig hearts when assessing infarct size with SPECT in pigs subjected to endovascular hypothermia. In the RAPID MI-ICE trial, we observed that several infarctions were patchy in appearance in the hypothermia group (Gotberg et al., 2010). The pattern differed from the classical wavefront phenomenon in infarct progression previously described by Reimer and Jennings (Reimer et al., 1977). The long-term significance of the observed patchy infarction pattern is not known. It could be speculated that the patchy pattern may induce reentry loops triggering arrhythmia, but on the other hand, the ejection fraction is maintained in salvaged myocardium, and reduced ejection fraction is the most important factor for development of ventricular arrhythmia.
The RAPID MI-ICE Clinical Trial
After seeing the rapid cooling effects of the combination of cold saline and endovascular cooling in our pig model, we investigated whether this combination could increase the number of patients achieving a temperature <35°C before reperfusion. In the COOL-MI and ICE-IT studies, only ∼30% reached <35°C before reperfusion (26% in anterior infarcts in COOL-MI, and 34% in ICE-IT). At the same time, we and other colleagues were concerned that administering a large volume of cold saline, up to 2 L, to patients with large infarctions might cause acute heart failure or pulmonary congestion. We therefore designed the RAPID MI-ICE clinical trial as a safety and feasibility study on 20 patients with large STEMI treated with primary PCI (Gotberg et al., 2010). Cardiac MRI was used for determination of infarct size and area at risk. The study showed that hypothermia was induced more rapidly with a combination of a rapid saline infusion together with endovascular cooling (Gotberg et al., 2010). All patients in the PCI+hypothermia group reached a temperature <35°C before reperfusion. This trial demonstrated that rapidly induced hypothermia was feasible and safe, and that the size of the infarct was significantly reduced (by 38% of area at risk) (Gotberg et al., 2010). Overall, hypothermia was well tolerated and troponin levels were also significantly reduced. Reperfusion was only delayed by 3 minutes, which is the time required for an experienced interventionist to insert an endovascular catheter into the vena cava via the femoral vein.
We were concerned that the infused volume of average 1.5 L cold saline could induce heart failure or pulmonary congestion. However, there was a trend toward fewer incidences of heart failure in the PCI+hypothermia group and no episodes of pulmonary congestion. There was a nonsignificant trend toward more episodes of pneumonia. It is well known that hypothermia increases the risk of infection by inhibiting the immune system, and the prolonged postreperfusion hypothermia period of 3 hours plus 3 hours of rewarming could increase the risk of lung infections as a result of immobilization and prolonged sedation of the patient. Shorter cooling duration could therefore be of advantage.
Analysis of the pooled results of the ICE-IT and RAPID MI-ICE trials
From the previous discussion, three important questions can be identified, which must be taken into consideration in the design of future trials.
1. Is it necessary to achieve a core body temperature of <35°C before reperfusion? 2. Is there a difference in the effect of hypothermia on anterior and inferior infarcts? 3. Is there a difference in the effect of hypothermia on infarcts with long and short durations of symptoms prior to reperfusion?
We performed a pooled post hoc analysis of the results obtained in the two clinical trials, ICE-IT and RAPID MI-ICE. The same endovascular cooling catheter (InnerCool RTx; Philips Healthcare), as we planned to use in the CHILL MI study, was used in these trials. In an effort to answer the specific questions just outlined, only patients treated per protocol, and who had undergone infarct size determination with SPECT or cardiac MRI, were included in the analysis.
Compared with the controls (i.e., those receiving PCI only, n=103), the group also subjected to hypothermia (n=94) showed a significant 24% reduction in infarct size, expressed as a percentage of the area of the LV myocardium (p<0.05, Erlinge et al., submitted). Among the hypothermia-treated patients in whom a core temperature below 35°C was achieved before reperfusion, the infarct size was reduced by 37% (p=0.01). The benefit observed was similar for both anterior (33% reduction; p=0.03) and inferior infarcts (42% reduction; p=0.04). Although not significant, a similar trend was seen in patients treated with hypothermia when divided according to duration of symptoms:<4 hours (reduced by 19%, p=0.15), and 4–6 hours (reduced by 33%, p=0.07).
The following conclusions can thus be drawn from the analysis of the pooled results:
1. Reaching a temperature of <35°C before reperfusion is of paramount importance in reducing the size of the infarct in the treatment of STEMI patients. 2. Hypothermia gives similar benefit in anterior and inferior infarcts. 3. Hypothermia seems to be beneficial up to 6 hours from the onset of symptoms.
Ongoing Clinical Trials
As a follow-up to COOL-MI, Radiant started COOL-MI 2 using a larger endovascular catheter to provide more rapid cooling. Hypothermia treatment was also started earlier in the emergency room to reach a temperature below 35°C before reperfusion. However, the study was terminated due to insufficient funding after 40 patients of the expected 225 had been treated. A trend toward reduced creatinine-kinase MB (CK-MB) has been reported and the results will be published in the near future.
Automated peritoneal lavage with the Velomedix system with cold saline is being used in the CAMARO trial. A European feasibility trial is ongoing with the aim of recruiting 20–100 STEMI patients and applying hypothermia for 3–12 hours (ClinicalTrials.gov identifier: NCT01016236). A corkscrew-like device is used to make a hole in the abdomen close to the navel of the conscious patient. However, bleeding could be a problem in STEMI patients, as they are normally heavily treated with anticoagulants and platelet inhibitors.
The Target Temperature Management trial is a randomized clinical trial with target temperatures of 33°C and 36°C for patients with cardiac arrest to limit cerebral injury; the cooling method is optional (ClinicalTrials.gov identifier: NCT01020916). The primary endpoint is all-cause mortality, but a substudy is being performed in which infarct size is being determined in relation to the area at risk using cardiac MRI, in patients with STEMI as the underlying cause of cardiac arrest. The number of patients expected in the substudy is planned to be 40–50. Previous post hoc analysis of a subset of 111 patients with cardiac arrest and STEMI in the Hypothermia After Cardiac Arrest (HACA) trial showed reduced CK-MB levels in patients who achieved the target temperature within 8 hours of cardiac arrest, but not after 8 hours (Koreny et al., 2009).
The CHILL-MI trial
Based on our animal experiments with the pig model and the RAPID MI-ICE safety and feasibility trial, we concluded that a combination of rapid endovascular cooling and cold saline infusion is a possible way of achieving therapeutic temperatures before reperfusion, with only little delay in reperfusion (3 minutes).
Based on these findings we designed and recently started the CHILL-MI trial (ClinicalTrials.gov identifier: NCT01379261) using endovascular cooling combined with rapid infusion of up to 2 L cold saline, including both anterior and large inferior STEMI, with up to 6 hours of symptoms prior to inclusion. The trial will include 120 patients at 10 international hospitals, and the myocardium at risk and infarct size will be measured after 4 days and 6 months with cardiac MRI. Hypothermia will be maintained for only 1 hour after reperfusion, followed by spontaneous rewarming.
One of the advantages of this short cooling protocol is that the whole cooling procedure can be performed in the PCI department, and the endovascular catheter can be removed before the patient is returned to the coronary care unit, thereby excluding the need for treatment in expensive and overcrowded intensive care units. Other advantages of short cooling postreperfusion are improved patient comfort and possibly a reduced risk of pulmonary infections. Smaller trials using variations of the CHILL-MI protocol are underway in Graz, Austria, and Albany (Table 3).
PCI, percutaneous coronary intervention; TTM, target temperature management.
Cooling the Conscious Patient
Most clinical experience of hypothermia has been obtained from cooling unconscious, anesthetized, and ventilated patients, either during surgery or after cardiac arrest. Cooling in these situations is more rapid and easier to maintain, because the rewarming/shivering response is abolished. Additional muscle relaxants can be given if required to totally obliterate shivering. In fact, the body temperature of these patients falls spontaneously. Cooling the conscious patient is much more difficult. As the body temperature falls, the temperature-controlling center in the brain stem triggers thermoregulatory defenses and powerful rewarming mechanisms. Skin circulation is reduced to maintain core temperature and shivering is initiated. There seems to be a shivering threshold at around 35°C–36°C, where shivering is most strongly activated. Below this temperature shivering becomes less intense. If shivering has become fully activated, it is almost impossible to cool the patient. The most important background factors determining the speed of cooling of the conscious patient is age and weight. Elderly patients have a much weaker shivering response, which facilitates cooling, while overweight patients have a larger body mass, which takes longer to cool.
Experience has shown that there are ways of fooling the body's temperature sensors. Most of the temperature sensors are located in the skin and are strong triggers of shivering. Therefore, surface cooling should be avoided in the conscious patient. When using endovascular cooling and intravenous saline infusions, it is possible to fool the skin's temperature sensors by surface counter warming, either with blankets or more advanced surface warming, such as a Bair Hugger®. A 4°C increase in skin temperature compensates for a 1°C decrease in core temperature for the shivering threshold (Cheng et al., 1995). A conscious patient undergoing endovascular cooling combined with surface warming does not feel cold, although their core temperature is 33°C.
Some medication is effective in reducing shivering, especially the kappa opioid receptor agonist meperidine (pethidine and demerol). Replacing morphine with meperidine in patients with AMI is very effective (10 mg meperidine is approximately equipotent to 1 mg morphine for pain relief). Meperidine has an almost instant effect on shivering, given in bolus doses of 25–50 mg. Buspirone is a serotonin 1A receptor partial agonist that is given as a tablet (usually 30 mg) and potentiates the antishivering effect of meperidine (Mokhtarani et al., 2001). Other suggested pharmaceutical treatments, such as morphine or alpha-2-receptor agonists, are less effective and it is doubtful whether magnesium has any effect at all.
Mechanisms of Action of the Cardioprotective Effect of Hypothermia
Much remains to be learned about the mechanisms by which hypothermia protects the heart from ischemia and reperfusion injury. The cardioprotective effect of hypothermia includes a reduction in metabolic demand with preserved ATP and glycogen stores (Badeer, 1956; Ohta et al., 1995; Simkhovich et al., 2004). This leads to a reduction in phosphate and glucose utilization in the cardiomyocytes (Jones et al., 1982; Ning et al., 1998; Simkhovich et al., 2004). Further, lactate accumulation is reduced (Ichihara et al., 1981). The systemic metabolism is also reduced, resulting in reduced demand for cardiac output, which reduces the workload on the heart. However, hypothermia has many more effects than simply reducing metabolism (Table 2). It reduces calcium and sodium overload during ischemia reperfusion injury in the heart (Anderson et al., 2006), and the release of reactive oxygen species at upon reperfusion is reduced (Gambert et al., 2004; Riess et al., 2004; Khaliulin et al., 2007). Mild hypothermia inhibits calcium-induced mitochondrial permeability transition pore (mPTP opening, which is a crucial apoptotic mechanism in ischemia reperfusion injury) (Tissier et al., 2009).
Hypothermia preserves myocardial function, promotes signaling for cell survival, inhibits apoptotic pathways, and reduces apoptosis (Tissier et al., 2011a; Ning et al., 2002, 2007; Huang et al., 2009; Meybohm et al., 2009). It reduces reactive hyperemia, that is, overcompensation of the coronary blood flow during the first few minutes of reperfusion (Olivecrona et al., 2007). It improves resistance to subsequent ischemia in the cardioplegic-arrested heart via the production of mRNA for heat shock protein 70–1 and mitochondrial proteins, adenine nucleotide translocator 1, and beta-F1-ATPase (Ning et al., 1999). Hypothermia has positive effects on several cardioprotective signal transduction pathways, including the cell survival protein p53 and hypoxia-inducible factor 1 (Ning et al., 2007). However, several additional mechanisms have been described (Polderman, 2009). Hypothermia mediates cardioprotection through the reperfusion injury salvage kinase pathway (PI3, Akt, NOS, and ERK), demonstrated by effects on Akt and the generation of nitric oxide (NO) (Shao et al., 2010). Further, Yang and coworkers (2011) recently found that the preservation of ERK activity during ischemia is involved in hypothermic cardioprotection. The protective protein kinase C isoform epsilon and protein kinase A are activated by hypothermia (Khaliulin et al., 2007; Khaliulin et al., 2010, 2011). Hypothermia also reduces the release of tissue-plasminogen-activating factor from the coronary endothelium, which is macrophage activating and has been shown to mediate reperfusion injury in the brain (van der Pals et al., 2009). Although hypothermia can mediate its cardioprotective effects via many pathways, some mechanisms are not involved. Bradycardia does not seem to be involved, as mentioned earlier (Chien et al., 1994), and it was recently shown that adenosine and opioid receptors do not mediate the cardioprotective effect of mild hypothermia (Darbera et al., 2011).
In summary, it is possible that the strong, highly reproducible cardioprotective effects of hypothermia are explained by its wide-ranging effects on most of the mechanisms involved in ischemia and reperfusion injury, and that the failure of other approaches in clinical trials could be explained by their highly selective effects on just one mechanism, leaving the other pathways unaffected.
Hemodynamic Effects of Hypothermia
Deep hypothermia (<30°C) decreases myocardial blood flow, reduces ventricular function, and causes spontaneous fibrillation (Tveita et al., 1994). However, these problems are not apparent during mild hypothermia. Clinical experience shows that cardiac output decreases during mild hypothermia, but this is caused by a reduction in systemic metabolism and the associated reduced demand for cardiac output. At 32°C, cardiac output is decreased by 30%–40%, but the metabolism is reduced by 50%–65%, which gives a net improvement in the balance between supply and demand (Polderman, 2009). Hypothermia causes a decrease in heart rate, most often resulting in a stable heart rate of 50–60 beats per minute. In the normal beating heart, mild hypothermia exerts a positive inotropic effect both in vitro (Suga et al., 1988; Kusuoka et al., 1991; Weisser et al., 2001) and in vivo (Nishimura et al., 2005; Post et al., 2010), but may cause mild diastolic dysfunction (Weisser et al., 2001; Schwarzl et al., 2011) (Table 1). Indeed, hypothermia may even improve hemodynamics in acute cardiogenic shock (see below) (Gotberg et al., 2010).
Myocardial perfusion is improved during mild core hypothermia in conscious humans (Frank et al., 2003). The peripheral resistance, especially in the skin circulation, increases. Generally, the mean arterial pressure is unchanged, while there is a slight reduction in systolic blood pressure, accompanied by a slight increase in diastolic blood pressure. The atrial natriuretic levels increase, antidiuretic hormone levels decrease, and sodium absorption in the kidney decreases resulting in a “cold diuresis,” which if not remedied will result in hypovolemia (Polderman, 2009). However, overall mild hypothermia usually stabilizes the circulation.
Side Effects of Hypothermia
Immunosuppression
Hypothermia causes immunosuppression by impairing neutrophil and macrophage function and leukocyte migration (Polderman, 2009). The risk of pneumonia may increase, as was seen in the stroke trial ICTUS (Hemmen et al., 2010). However, in the HACA and Bernard trials on cardiac arrest, no increase in infection was seen in the hypothermia groups (Bernard et al., 2002; 2002).
Coagulation and platelet function
The coagulation system is inhibited by hypothermia, at least at temperatures <34°C (Polderman, 2009). It has been suggested that platelets are also inhibited by hypothermia, and there are some studies supporting this notion (Michelson et al., 1994, 1999; Frelinger et al., 2003). However, there are more recent reports suggesting increased platelet reactivity during mild hypothermia (Zhang et al., 2004; Lindenblatt et al., 2005; Scharbert et al., 2006; Xavier et al., 2007). We examined platelet activity at 33°C and 37°C and found unchanged or slightly increased platelet activity for most agonists (Hogberg et al., 2009). Interestingly, a resistance to the adenosine diphosphate (ADP) inhibitor clopidogrel was induced, possibly due to increased sensitivity to ADP activation (Hogberg et al., 2009).
The treatment of cardiac arrest patients with hypothermia in the clinical setting has not revealed bleeding complications to be a major problem. Our recommendation for treating cooled STEMI patients is to be very observant that not to overdose anticoagulants such as heparins or bivalirudin, and to give antiplatelet therapy as usual.
Metabolism
Increased fat metabolism leads to increased levels of glycerol, free fatty acids, and lactate in the blood, which cause mild metabolic acidosis during long periods of hypothermia. Insulin secretion is reduced and together with a relative insulin resistance this may result in increased glucose levels (Lehot et al., 1992; Polderman, 2009).
Ion changes
Hypothermia induces hypokalemia after longer periods of treatment (Polderman, 2009). It is important not to overcompensate this with potassium infusions, since the condition will be reversed upon rewarming.
Arrhythmia
Electrocardiographic changes induced by hypothermia include prolonged PR intervals, increased QT intervals, and widening of the QRS complex (Polderman, 2009). Mild hypothermia does not increase the risk of arrhythmia. Indeed, mild hypothermia increases membrane stability and decreases the risk of arrhythmia. However, more severe hypothermia, below 28°C, can increase the risk of arrhythmia and cause spontaneous ventricular fibrillation. In addition, such arrhythmia is more difficult to treat, as the myocardium is less responsive to antiarrhythmic drugs. In contrast, the risk of ventricular fibrillation is reduced during mild hypothermia by the stabilization of cardiomyocyte membranes (Harada et al., 2008). Mild hypothermia (33°C) has been shown to improve the success of defibrillation compared with normothermia, with an increased chance of achieving return of spontaneous circulation (ROSC) (Boddicker et al., 2005). More severe hypothermia (30°C) has been found to facilitate transthoracic defibrillation in a swine model, while moderate hypothermia (33°C) did not alter the energy required for defibrillation (Rhee et al., 2005). Since impedance increases and current falls during hypothermia, the improved shock success is due to a hypothermia-induced change in the mechanical or electrophysiological properties of the myocardium (Rhee et al., 2005).
Cardiogenic Shock
Even patients experiencing cardiogenic shock have a good outcome after treatment with hypothermia (Hovdenes et al., 2007). We examined the effects of hypothermia in a pig model of cardiogenic shock (Gotberg et al., 2010). Cooling one group of pigs to <34°C resulted in better survival; 5 out of 8 pigs in the normothermia group died, while all pigs in the hypothermia group survived (n=8). Stroke volume and blood pressure were maintained at a higher level in the hypothermia group, whereas the heart rate was significantly lower. Cardiac output did not differ between the groups. Blood gas analysis revealed higher mixed venous oxygen saturation, pH, and base excess in the hypothermia group, indicating less metabolic acidosis (Gotberg et al., 2010).
In a model of resuscitated pigs, Schwarzl and coworkers (2011) used pressure–volume analysis and found improved LV systolic function during mild hypothermia, but also decreased LV end-diastolic distensibility. Mild hypothermia did not increase plasma catecholamine levels, and spectral analysis of heart rate variability revealed reduced sympathetic activation. They concluded that mild hypothermia after cardiac resuscitation improves systolic myocardial function without further sympathetic activation. Reduced metabolism during hypothermia has been reported to outweigh the decrease in cardiac output and to thus act favorably on the balance between systemic oxygen supply and demand (Schwarzl et al., 2011).
In summary, hypothermia reduces acute mortality in cardiogenic shock models, improves hemodynamic parameters, and reduces metabolic acidosis. These findings suggest a possible clinical benefit of therapeutic hypothermia for patients with acute cardiogenic shock. This is of special interest in the treatment of STEMI patients, because cardiogenic shock is responsible for 50% of the mortality among STEMI patients.
Future Development
If hypothermia can be shown to be effective as an adjunctive treatment for myocardial infarction, it will open the way for other developments. More rapid cooling methods, such as liquid ventilation cooling, maybe developed for clinical use. Practical cooling methods that can be used in ambulances and at referral hospitals could allow hypothermia to be initiated earlier during ischemia, further reducing the extent of the infarct. Drugs that induce hypothermia have been suggested or are under development (haloperidol, cannabinoids, melatonin, neurotensin, transient receptor potential vanilloid 1 agonists [synthetic chili pepper], and xenon). Hypothermia could be clinically used to treat cardiogenic shock, which still has a mortality rate of nearly 50%, or to stabilize the circulation during septic shock. Rapid initiation of hypothermia in the field could improve ROSC in patients with cardiac arrest resulting from ventricular arrhythmia.
Conclusions
In conclusion, mild hypothermia may be of benefit as an adjunctive treatment for STEMI as it has positive effects on the four components of ischemia reperfusion injury: myocardial stunning, microvascular obstruction, reperfusion arrhythmia, and lethal reperfusion injury. To reduce cerebral injury after cardiac arrest, hypothermia is initiated after reperfusion and should be maintained for 24–48 hours. In contrast, for heart protection, evidence suggests that therapeutic hypothermia should be instigated as early as possible during ischemia, at least before reperfusion. Clinical results indicate that achieving a core temperature of <35°C before reperfusion is of paramount importance to reduce the extent of the infarct in the treatment of STEMI patients. Continued treatment after reperfusion can probably be relatively short. Hypothermia has wide-ranging effects on most of the mechanisms involved in ischemia and reperfusion injury, which may explain the potent, highly reproducible cardioprotective effects seen in a multitude of studies in different species. Cooling conscious patients with STEMI is safe, feasible, and well tolerated, but antishivering strategies must be employed. Larger clinical studies such as the CHILL-MI trial are ongoing and are needed to confirm the cardioprotective effect in man.
Funding
This study was supported by the Swedish Research Council, the Swedish Heart and Lung Foundation, and the Vascular Wall program (Faculty of Medicine, Lund University).
Footnotes
Disclosure Statement
The author received endovascular cooling catheters and funding for MRI examinations in the RAPID MI-ICE study from InnerCool Therapies, now Philips Healthcare.