
Abstract
Changes in the dynamic interactions of macromolecules in cell membranes appear to underlie the robust neuroprotective effect of hypothermia against selective neuronal degeneration in the CA1 region of the rat hippocampus after transient cerebral ischemia, but the detailed mechanisms are still elusive. Using the two-vessel occlusion model of transient normothermic cerebral ischemia of 15 min duration, we investigated the tyrosine phosphorylation of synaptic proteins in general and that of the NMDA receptor subunits in particular, at different times of recirculation. Specifically, the effect of intra-ischemic hypothermia (33°C), which provides neuroprotection to the CA1 region of the hippocampus, was studied. Phosphorylation of tyrosine residues on the NMDA receptor (NR) 2, but not of the NR1 or the AMPA receptor subunit 1 (GluR1) proteins, was markedly enhanced following cerebral ischemia. Protein tyrosine phosphorylation was persistently increased in the postsynaptic densities of the vulnerable CA1 region, but was transient in the CA3/dentate gyrus (DG) neurons where cell death was not evident. The phospho-tyrosine phosphatase activity decreased during reperfusion in the CA1 region but not in CA3/DG. Importantly, decreasing body temperature to 33°C during ischemia modified the dynamics of the protein tyrosine phosphorylation of NR2 in the CA1 region, which was transient and similar in time course to that seen in the CA3/DG region after normothermic ischemia. We conclude that the protracted tyrosine phosphorylation of the NR2 subunit in the hippocampus CA1 region following normothermic ischemia is attenuated by hypothermia and therefore constitutes an important target for hypothermic neuroprotection.
Introduction
Dysregulated cell signaling in the CA1 region in the postischemic phase is evident by the persistent increase in protein phosphorylation, particularly that of the NR2A subunit (Hu and Wieloch, 1994; Takagi et al., 1997; Takagi et al., 2010). This protracted change in cell signaling causes cellular malfunction that ultimately leads to cell death in a delayed fashion, including depression of protein synthesis, changes in gene expression, or activation of mitochondria induced cell death processes (Endres et al., 2008). In the present investigation, we demonstrate that intra-ischemic hypothermia prevents protracted tyrosine phosphorylation of NR2 proteins in the CA1 region during recovery following global ischemia, implicating this process as target for hypothermic neuroprotection.
Materials and Methods
Surgical procedures
Normothermic and hypothermic ischemia was employed, using the two-vessel occlusion model of global cerebral ischemia (Smith et al., 1984). The animal experiments were approved by the ethical committee at Lund University. Male Wistar rats (350–400 g; Møllegaard A/S, Denmark) were used. The animals were fasted overnight. Anesthesia was induced with 2% halothane oxygen/nitrous oxide (30%/70%). The anesthetized rats were intubated and artificially ventilated. An external jugular vein catheter, a tail artery catheter, and one vein catheter were also inserted. The arterial blood pressure was measured and recorded. An incision was made in the neck, and both common carotid arteries were exposed and encircled by loose ligatures. Blood gases and blood glucose was measured, and were in the following range: PaO2 >90 mmHg, PaCO2 35–45 mmHg, pH 7.35–7.45. Bipolar EEG electrodes were inserted into the temporal muscles, and the EEG activity was recorded. The inspired halothane concentration was decreased to 0.3%, and 150 IU kg−1 heparin was administered i.v. Vecuronium (Organon, Teknika, Boxtel, Holland), a muscle relaxant, was administered i.v. (0.7 mg followed by an infusion of 3 mg h−1). A steady state was maintained for 30 min prior to the induction of ischemia. Blood was withdrawn through the jugular catheter in order to reach a mean arterial blood pressure (MABP) of 50 mmHg and both carotid arteries were clamped. MABP was maintained at 50 mmHg during the ischemic period (15 min) by withdrawing or infusing blood through the jugular catheter. Ischemia was defined as the time of the onset of isoelectric EEG at an MABP of 50 mmHg. Ischemia was terminated by removal of the clamps and reinfusion of blood through the jugular catheter, followed by 0.5 ml of 0.6 M sodium bicarbonate. Scull and rectal temperature (kept at approximately 37°C) was monitored and controlled with a heating pad. At the end of ischemia, Vecuronium and halothane were discontinued, the external jugular vein catheter removed, and all wounds sutured. Within 45 min of reperfusion, the animals breathed spontaneously and the endotracheal tube was removed. The brains (n=4 per time point) of the anesthetized animals were freeze-fixed in situ with liquid nitrogen at 30 min, 4 h, and 24 h of recovery after ischemia. Following the removal of the frozen brain from the scull, the CA1 and the CA3/DG (dentate gyrus) regions were dissected from dorsal hippocampus, and the neocortex was sampled at the same hippocampal level.
Hypothermic ischemia was performed by covering the body with ice until a rectal and subdural temperature of 33 °C was reached prior to the induction of ischemia. A lamp was used to normalize the body temperature of the hypothermic animals after the insult. Normothermia was reached within 15 min of the start of heating.
Subcellular fractionation
The tissue (5–20 mg) was sonicated 2×10 s in 1:10 (mg/μl) homogenization buffer consisting of 50 mM MOPS pH 7.4; 2 mM dithiothreitol; 100 mM KCl; 0.5 mM magnesium chloride; 1 mM sodium orthovanadate; 0.1 mM EDTA; 50 μg/ml leupeptin; 10 μg/ml aprotinin; 5 μg/ml pepstatin; and 0.32 M sucrose. The homogenates were spun at 800g for 10 min at 4°C followed by centrifugation of the supernatant at 9200g for 15 min at 4°C in a SE12 rotor of Sorvall. The resulting pellet (P2) was collected by sonication of 10 s in homogenization buffer containing 0.1% Triton X-100, and the supernatant was spun at 165,000g for 1 h at 2°C in a TL100.2 rotor. The supernatant (S3) fraction, consisting of the cytosol, and the particulate fraction (microsomal fraction or P3 fraction) reconstituted in homogenization buffer plus 1% Triton X-100 were either separated on sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) or frozen at −80°C for later analysis.
For preparation of synaptic vesicles, synaptosol and synaptic junctions, the tissue was homogenized in 10 strokes in 1:5 (mg/ml) homogenization buffer with Teflon-glass homogenizer. The P2 fraction, the pellet between 800g and 9200g, was washed twice with homogenization buffer without detergent and centrifuged at 10,200g for 15 min at 4°C. The pellet was lysed by hypo-osmotic shock in nine volumes of 5 mM HEPES/NaOH (pH 8.4) and stirred on ice for 30 min. The lysate was sonicated for 2×10 s with a cooled sonication probe in liquid nitrogen and centrifuged at 25,000g for 20 min at 4°C to yield the lysate supernatant and the lysate pellet. The obtained pellet was resuspended in homogenization buffer containing 0.32 M sucrose, layered onto a discontinuous sucrose gradient of 1 ml of each 0.8 M/1.0 M/1.2 M, and centrifuged at 65,000g in a TL100.3 rotor for 2 h at 4°C. A crude postsynaptic density fraction at the 1.0 M/1.2 M interface was collected and washed in five volumes of the homogenization buffer and collected by centrifugation.
A more pure fraction of postsynaptic densities (PSDs) from neocortex was obtained from six rat brains by a similar procedure as above except that a sucrose gradient of 1.0/1.5/2.0 M sucrose was utilized in the last centrifugation. The synaptosomal membrane was collected from the sample/1.0 M sucrose interface. The PSDs pellet were obtained by diluting the layer between 1.5 M and 2.0 M sucrose interface with equal volume of water and 1% Triton X100/150 mM KCl and spinning at 275,000g for 1 h.
Deglycosylation was conducted using Endoglycosidase F/N-glycosidase F according to the supplier (Boehringer Mannheim Biochemica, Mannheim, Germany). The PSDs were denatured in the presence of 1% SDS and 0.5% (W/V) n-octylglucoside in a sodium phosphate buffer containing 10 mM EDTA and 10 mM sodium azide. Immunoprecipitation of NR1 and NR2 was performed under denaturating conditions. The PSD fraction (75 μg) were incubated with antibodies (2.0 μg) against NR1 or NR2 (Millipore, Temecula, CA) overnight. Pansorbin (Calbiochem, La Jolla, CA), prepared according to protocol of Transduction Laboratories (Lexington, KY), was used for collecting the immuno-complexes.
Electrophoresis
Electrophoresis was carried out on 10% SDS-PAGE. Antibodies against phosphotyrosine (Affinity polyclonal, PY20, and Zymed monoclonal and polyclonal antibodies) were primary antibodies and a horseradish peroxidase labeled anti-rabbit antibody was the secondary antibody. Immunoblots were developed with the Amersham ECL™ system. The autoradiograms were scanned and quantified by computerized image analysis using Image J. 10 mM phosphotyrosine but not phosphoserine and phosphothreonine blocked immunoreactivity, when incubated with the anti-phosphotyrosine (Ptyr) antibody and the Immobilon-P membrane. The glutamate receptor antibodies were from Chemicon.
Protein tyrosine phosphatase activity measurements
The PTPase activity was determined (Brautigan and Pinault, 1991) by mixing 5 μl of 1 μg/μl crude PSD fraction, 5 μl of 20 μg/ml [γ-32P] MBP solution, and 20 μl assay buffer consisting of 25 mM HEPES (pH 7.0), 1 mM EDTA, 0.1 mg/ml BSA, 30 mM 2-mercaptoethanol were mixed and incubated at 30°C for 10 min. At the end of incubation, 2 μl of mixture was sampled for measuring total radioactivity, 60 μl of cold 10% TCA was added to the residual mixture, and the precipitate was spun down. The radioactivity in the supernatant was counted and the extent of dephosphorylation was calculated.
Results
Phosphorylation of the NMDA receptor subunit 2
In isolated post-synaptic densities from the cortex of sham-operated rats and rats exposed to 15 min of transient cerebral ischemia with 4 h of reperfusion, a Ptyr protein with an apparent molecular weight of 180 kDa (PSD-180) was detected (Fig. 1A). The PSD-180 band was enriched in PSD fraction compared to the synaptic membrane fraction (SM), and its phosphorylation was notably enhanced during reperfusion. High levels of NR1 (Fig. 1B) and NR2A/B subunits (Fig. 1C) were also found in the PSD fraction, but their levels did not change during recovery. The glutamate receptor subunits are glycoproteins, and deglycosylation changes their electrophoretic mobility. The PSD-180 Ptyr protein was identified as NR2, by first treating samples with endoglycosidase F/N-glycosidase F in order to deglycosylate the receptor proteins. Endoglycosidase F/N-glycosidase F shifted the mobility of PSD-180 from 180 kDa to 160 kDa, which matched the shift of NR2 (Fig. 2A). Immuno-precipitation of NR2A/B followed by electrophoresis and Western blotting, and with subsequent labeling with the anti-NR2A/B and anti-Ptyr antibodies (Fig. 2B), confirmed that NR2A/B are phosphorylated on tyrosines following ischemia and correspond to PSD-180. The blot of the NR2A/B immunoprecipitate was labeled by both an anti-NR2A/B antibody and an anti-Ptyr antibody as a single band at the 180 kDa position. The level of anti-NR2 was unchanged, while tyrosine phosphorylation clearly increased at 4 h of reperfusion. The PSD-180 in postischemic brains is therefore identified as the NR2 subunit, in accordance with earlier observations showing that the NR2A subunit is a major phosphotyrosine (Ptyr) protein of PSDs (Takagi et al., 1997).
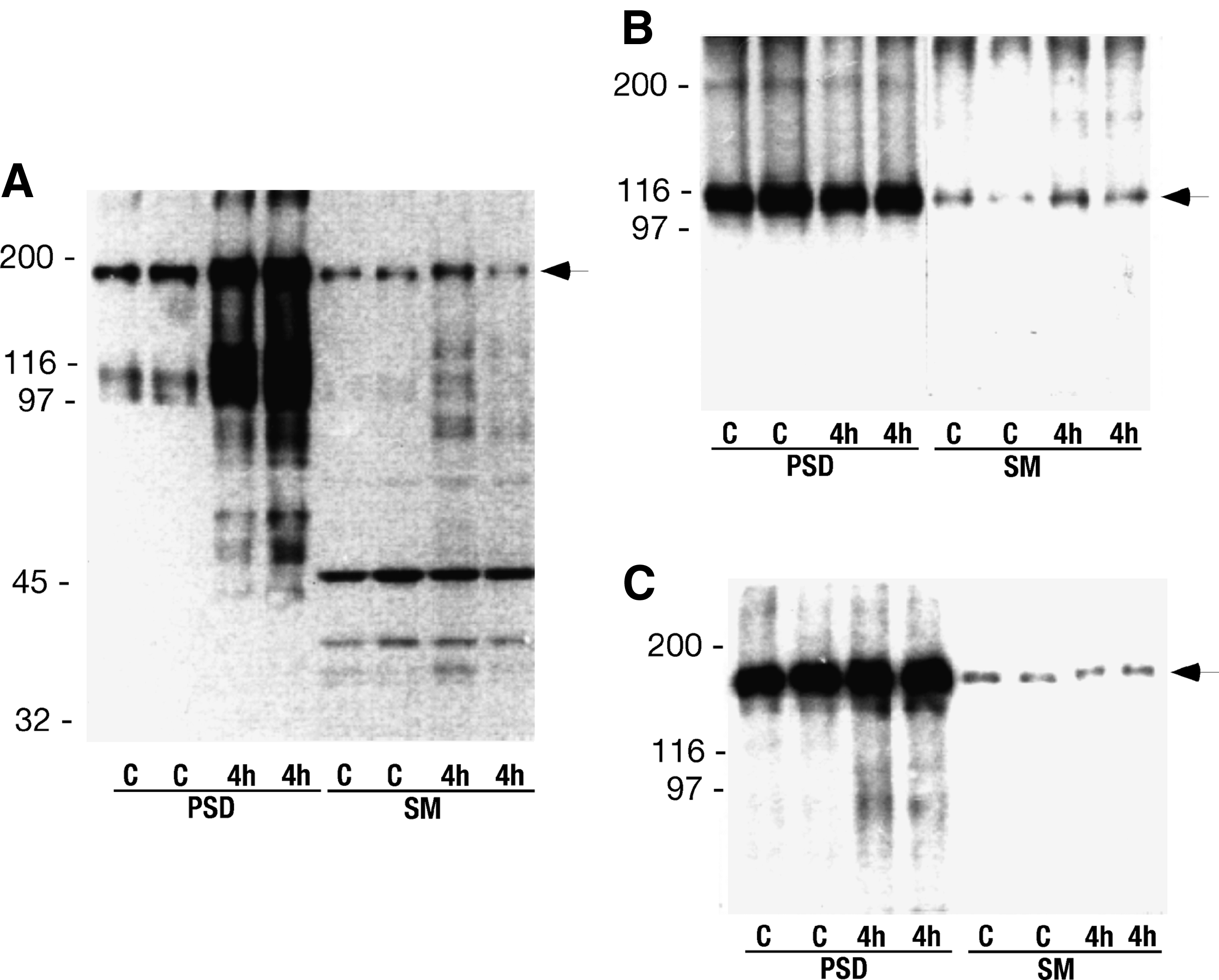
Autoradiograms showing tyrosine phosphorylation of a 180 kDa protein (arrow) (

Identification of tyrosine phosphorylated NMDA receptor subunits. (
Deglycosylation changed the mobility of NR1 and GluR1 (Fig. 2C), but there was no corresponding Ptyr staining (Fig. 2A). Immunoprecipitated NR1 was also not phosphorylated on tyrosines (Fig. 2D) following ischemia.
The tyrosine phosphorylation of NR2 is mitigated by hypothermia
To assess the relevance of the tyrosine phosphorylation of the NR2 subunits for hypothermic neuroprotection, its phosphorylation state was assessed in the CA1 and CA3/DG regions, areas that are differentially sensitive to global ischemia. The alterations in tyrosine phosphorylation of proteins in the crude PSD fractions up to 24 h of recovery following ischemia under normothermic (37°C rectal temperature) and hypothermic (33°C) conditions is shown in Figure 3A and B. Under normothermic conditions, there was a continuous enhancement of tyrosine phosphorylation in the sensitive CA1 region, while in areas where degeneration does not occur (CA3/DG), the phosphorylation state peaked at 4 h of reperfusion and then subsided. Following hypothermic global ischemia, tyrosine phosphorylation still increased during reperfusion, but in the CA1 region where hypothermia provides protection phosphorylation was transient diminishing at 24 h compared to the 4 h time point. In the CA/DG region phosphorylation was generally diminished and by 24 h not different from pre-ischemic levels. Although there was an increase in PTyr immunoreactivity of the NR2 bands, there were no changes in the levels of NR2A/B protein, as indicated by Western blot analysis (Fig. 3C).
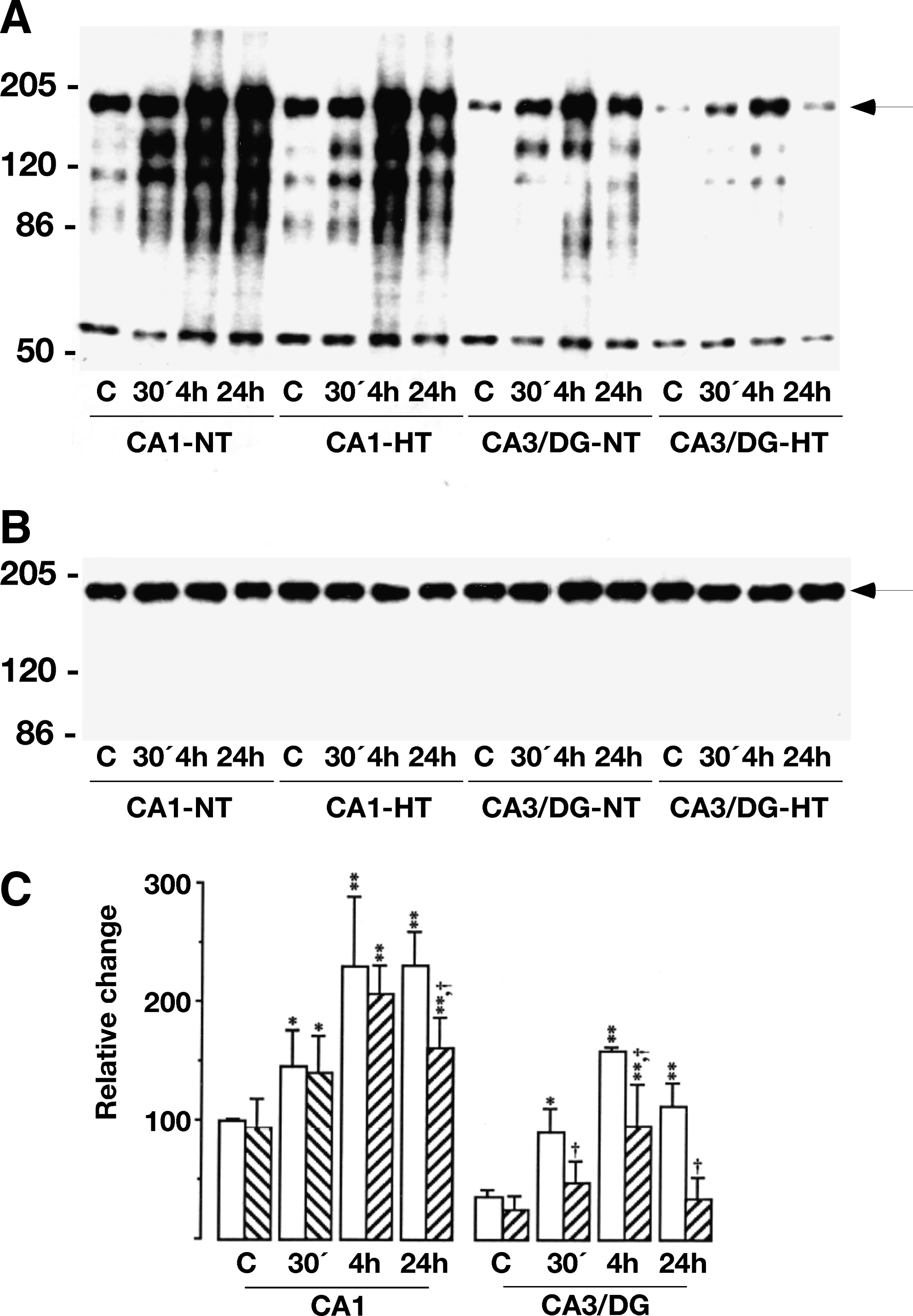
The dynamic changes in the tyrosine phosphorylation of proteins in the crude PSD fraction from hippocampal CA1 and CA3/dentate gyrus (DG) regions following transient cerebral ischemia. The tissue was from normothermic (NT, 37°C) and hypothermic (HT, 33°C) sham-operated rats and animals exposed to 15 min ischemia and 30 min (30), 4 h, and 24 h of reperfusion. (
Since the phosphorylation state of proteins reflects the balance between protein kinase and phosphatase activities, we examined the total PTPase activity in the crude PSD fractions from the CA1 and CA3/DG regions (Table 1). The tyrosine phosphatase activity decreased significantly by 28±18% at 6 h of reperfusion in the CA1 region, while the activity was unaffected in the CA3/DG region.
Values are means±SD (n=4). *p<0.05 (Student's t-test) denotes significant difference from sham-operated control group (100% – value).
Statistical analysis
Statistical analysis was performed using ANOVA with a post hoc Scheffee's test with p<0.05 denoting a significant difference.
Discussion
In the present investigation, we demonstrate a protracted tyrosine phosphorylation of a 180 kD protein in the PSDs of rat neocortex and hippocampus, which we identified as the NR2 subunit, in accordance with earlier findings (Takagi et al., 1997). We also demonstrate that protein tyrosine protein phosphorylation of NR2 in the relatively ischemia-resistant CA3/DG region was transient over the 24 h of reperfusion. Furthermore, the increase in tyrosine phosphorylation in CA1 but not in the CA3 region was associated with a decrease in PTPase activity. Importantly, following global ischemia of 15 min duration with a body temperature of 33°C, tyrosine phosphorylation in the CA1 region is transient and the time course of phosphorylation is similar as that seen in the resistant CA3/DG region after ischemia conducted at 37°C. Hence, in cells that succumb after global ischemia, tyrosine phosphorylation of NR2 is persistently enhanced, while in surviving neurons the phosphorylation is transient. This implies that tyrosine kinases are transiently dysregulated in surviving neurons, and that protracted phosphorylation occurs in cell destined to die after ischemia.
How does this come about? Tyrosine phosphorylation regulates synaptic activity and efficacy. In particular, the Src family kinases (SFK) are important in the regulation of NMDA receptor function by tyrosine phosphorylation (Salter and Kalia, 2004). The SFKs are integral parts of the biomolecular complexes forming protein superstructures in the PSD with receptor subunits including those of the NMDA receptor (Traynelis et al., 2010), such as the NR2A-PSD95-Src complex (Salter and Kalia, 2004). In PSDs, tyrosine kinase inhibitors depress and tyrosine phosphatase inhibitors enhance NMDA receptor-mediated calcium currents. Therefore, under physiological conditions, regulation of tyrosine phosphorylation of NR2 changes synaptic efficacy and receptor conductance (Salter and Kalia, 2004; Traynelis et al., 2010). After cerebral ischemia, the SFKs are activated (Hu et al., 1998; Takagi et al., 1999), explaining the marked tyrosine phosphorylation that persists, at least during 24 h of recovery and essentially until neurons degenerate, which implies that excessive and protracted tyrosine phosphorylation of NR2 may contribute to neuronal damage. The latter is supported by findings that suppression of PSD-95 expression attenuates the phosphorylation of NR2A and its interaction with Src and Fyn and is neuroprotective (Hou et al., 2003; Wang et al., 2010). Also, an inhibitor of SFKs protects against ischemic damage in the hippocampus (Hou et al., 2007). It appears that the enduring phosphorylation of the NR2A through the NR2A-PSD95-Src singalosome leads to an imbalance in cell signaling which induces ischemic damage or apoptosis (Wieloch et al., 1996; Miyawaki et al., 2009). Also, toxic exposure of neurons in the CA1 region to Aβ 25–35 induces tyrosine phosphorylation of NR2A (Wu and Hou, 2010). However, tyrosine phosphorylation per se is not detrimental, since this is seen in the resistant CA3 regions in the present study, and in the CA1 region following preconditioning (Shamloo and Wieloch, 1999), albeit in these instances phosphorylation is transient.
What causes the persistent phosphorylation of NR2? We found a selective decrease in PTPase activity occurring in the CA1 region but not in the CA3 region, which suggests that increased tyrosine phosphorylation could in addition to activation of SFKase be due to a decrease in the PTPase activity (Table 1). It was recently demonstrated that calpain mediates degradation of PTPase under oxidizing conditions (Trümpler et al., 2009), which could occur after ischemia (Endres et al., 2008). We envisaged that tyrosine phosphorylation of NR2 in CA1 is a combination of ischemia-induced activation of the SFKs and a depression of PTP activity, while in the CA3/DG region reversible activation of SFKs occurs.
Evidently, intra-ischemic hypothermia prevents the protracted dysregulation of NR2 tyrosine phosphorylation. During ischemia and reperfusion, dramatic structural changes and molecular rearrangements occur in synapses. In cell cultures subjected to oxygen and glucose deprivation, spines collapse and synaptic actin polymers dissolves and form dendritic aggregates (Gisselsson et al., 2005). At the molecular levels, ischemia induces irreversible movement, translocation, of regulatory proteins such as protein kinase C or Calcium Calmodulin kinase II (CaMKII) from cytosol to cell membranes and PSDs (Cardell et al., 1991; Hu et al., 1995, 1998). Interestingly, 80–90% of Src and 50–60% of NMDA receptor subunits are associated with lipid rafts after ischemia (Besshoh et al., 2005). Decreasing the temperature to 33°C during ischemia or hypoxia/aglycemia diminishes actin depolymerization, shortens spine length, dramatically decreases spine motility (Gisselsson et al., 2005), and prevents translocation of proteins to PSDs (Cardell et al., 1991; Hu et al., 1995). We propose that intra-ischemic hypothermia prevents lipid raft-mediated irreversible synaptic rearrangements and aggregation of proteins in the PSD, which causes protracted activation NR2A-PSD95-Src complex and subsequent tyrosine phosphorylation of NR2A.
We conclude that abnormal and persistent tyrosine phosphorylation of NR2 receptor subunits may contribute to dysregulated glutamatergic neurotransmission following brain ischemia, causing neuronal dysfunction and ultimately cell death, and that this process may be prevented by mild hypothermia. Also, the tyrosine kinases phosphorylating NR2 may be future targets for neuroprotective compounds.
Footnotes
Acknowledgments
This work was funded by the Swedish Medical Research Council (grant number 08644), the EU seventh work program through the European Stroke Network (201024), The Pia Ståhl Foundation, and The Swedish Brain Fund.
Disclosure Statement
TW is the founder of Quickcool AB, a company developing devices for body temperature management.