
Abstract
Interleukin-6 (IL-6) is a proinflammatory cytokine that may play multiple roles in the pathogenesis of traumatic brain injury (TBI). The present study determined time-dependent changes in IL-6 concentrations in vulnerable brain regions, cerebrospinal fluid (CSF) samples, and plasma after normothermic TBI. Because secondary insults are common in head injured patients, we also assessed the consequences of a post-traumatic secondary hypoxic insult on this pleiotropic cytokine. Male Sprague-Dawley rats were intubated, anesthetized, and underwent a moderate parasagittal fluid-percussion brain injury (1.8–2.1 atm, 37°C) followed by either 30 minutes of normoxic or hypoxic (pO2=30–40 mmHg) gas levels. Rats were sacrificed 3, 6, or 24 hours after TBI or sham-operated procedures. Brain samples, including the ipsilateral cerebral cortex and hippocampus were dissected and analyzed. Plasma and CSF samples were collected at similar times and stored at −80°C until analysis. IL-6 levels were significantly increased (p<0.05) at 3, 6, and 24 hours in the cerebral cortex and at 6 hours in the hippocampus after TBI. IL-6 levels in the TBI normoxic group for both structures returned to control levels by 24 hours. Plasma levels of IL-6 were elevated at all time points, while CSF levels were high at 3 and 6 hours, but normalized by 24 hours. Post-traumatic hypoxia led to significantly elevated (p<0.05) IL-6 protein levels in the cerebral cortex at 24 hours compared to sham-operated controls. These findings demonstrate that moderate TBI leads to an early increase in IL-6 brain, plasma, and CSF protein levels. Secondary post-traumatic hypoxia, a common secondary injury mechanism, led to prolonged elevations in plasma IL-6 levels that may participate in the pathophysiology of this complicated TBI model.
Introduction
In experimental models of cerebral ischemia/hypoxia or TBI, early and transient profiles of IL-6 elevations have been documented (Woodroofe et al., 1991; Taupin et al., 1993; Maeda et al., 1994; Wang et al., 1995; Hagberg et al., 1996; Bell et al., 1997; Bona et al., 1999; Stover et al., 2000; Clausen et al., 2011; Mukherjee et al., 2011). Commonly, maximum increases in brain IL-6 occur during the first several hours after injury, with normalization by 24 to 48 hours. In attempting to compare these experimental data to clinical findings, it is important to appreciate that head-injured patients commonly undergo multiple trauma or secondary insults, including hypotension or hypoxia resulting in multiple organ dysfunction (Chesnut et al., 1993; Bardenheuer et al., 2000; Rockswold et al., 2006; Probst et al., 2012). Thus, an important question is whether simple or more complicated models of experimental TBI are most appropriate to mimic the clinical condition and to investigate injury mechanisms and test novel therapeutic interventions (Matsushita et al., 2001; Statler et al., 2001). For example, in a recent study where animals were subjected to closed head injury, femoral fracture, and hemorrhagic shock, an increased inflammation, including elevated blood levels of IL-6 were reported compared to animals undergoing TBI alone (Probst et al., 2012). In clinical studies where secondary insults may be common, IL-6 has been reported to be an effective biomarker for brain injury severity and may be predictive for a delayed increase in intracranial pressure (Kim et al., 2000; Woiciechowsky et al., 2002; Hergenroeder et al., 2010).
Secondary hypoxia caused by impact apnea and respiratory distress are common occurrences after head injury. Secondary hypoxia has been reported to occur in 45.6% of severely head injured patients (Katsurada et al., 1973; Sinha etal., 1973; Chesnut et al., 1993). Experimental TBI studies have consistently reported the detrimental consequences of secondary hypoxia or impact apnea on traumatic outcome (Cherian et al., 1986; Ishige et al., 1987a, 1987b; Jenkins et al., 1989; Clark et al., 1997; Bramlett et al., 1999a, 1999b; Matsushita et al., 2001; Gao et al., 2010; Garman et al., 2011). In these investigations, secondary hypoxia was reported to worsen histopathological outcome, increase brain edema, decrease cerebral perfusion, and aggravate behavioral outcome. Although few experimental studies have assessed the effects of secondary insults on the neurochemical consequences of TBI, several experimental and clinical studies suggest that this is an important area of future investigation (Buttram et al., 2007; Frink et al., 2009; Probst et al., 2012; Weckbach et al., 2012).
The purpose of this study was to investigate whether experimentally induced TBI accompanied by hypoxia would alter the temporal profile of IL-6 protein levels. For this study, we utilized a TBI model followed by a secondary hypoxic insult that has been reported to significantly increase cortical contusion volume under normothermic conditions (Bramlett et al., 1999b). To this end, IL-6 levels were measured in vulnerable brain regions, CSF, and plasma samples at 3, 6, and 24 hours after injury.
Materials and Methods
Traumatic brain injury
Forty-five male Sprague-Dawley rats weighing between 270 and 399 g were used for this experiment. The animals were maintained on a 12-hour light/12-hour dark cycle and given food ad libitum. The protocol for this animal study was approved by the University of Miami Animal Care and Use Committee and all animal procedures followed guidelines published in the NIH Guide for the Care and Use of Laboratory Animals. Animals were anesthetized (1% halothane, 70/30% nitrous oxide/oxygen) 24 hours before trauma and surgically prepared for fluid-percussion (F-P) brain injury as described previously (Dietrich et al., 1994). Briefly, a 4.8-mm craniotomy was made overlying the right parietal cortex (3.8 mm posterior to the bregma and 2.5 mm lateral to the midline) (Zilles, 1985) and a plastic injury tube was placed over the exposed dura. The tube was bound by adhesive and dental acrylic, which was permitted to harden, and the scalp was then sutured. The animal was allowed to recover before being returned to the home cage.
Food was withheld overnight, and on the following day, a F-P device was used to produce experimental TBI (Dixon et al., 1987). Intubated, anesthetized (0.5% halothane, 70/30% nitrous oxide/oxygen) animals were subjected to a moderate F-P injury (1.87–2.17 atm). Before trauma, the femoral artery was cannulated to monitor blood gas levels. Brain temperature was measured with a probe in the temporalis muscle and body temperature via the rectal probe. Both brain and body temperature were maintained at normothermic (37°C) levels before and after TBI.
Secondary hypoxia
After receiving a moderate F-P injury, the animals were randomized and either maintained for 30 minutes at either normoxic (TBI-Normoxia, n=4–9) or hypoxic (TBI-Hypoxia, n=4–7; pO2=30–40 mmHg) gas levels as previously published (Bramlett et al., 1999a, 1999b). Hypoxia was induced by reducing the level of oxygen (11%) and increasing the level of nitrous oxide (56%) and the addition of nitrogen (33%) to the gas mixture. This results in a pO2 level ranging from 30 to 40 mmHg. Blood gases were measured 15 minutes before TBI and at 10, 30, and 45 minutes post-TBI. Sham animals (n=12) underwent all manipulations except for the actual trauma or hypoxic insult.
Enzyme-linked immunoabsorbent assay
IL-6 protein levels in sham-operated and traumatized rats were quantified using a commercially available enzyme-linked immunoabsorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) by an investigator blinded to the experimental groups. At 3, 6, or 24 hours after injury or sham surgery, rats were deeply anesthetized and plasma and CSF samples were obtained. CSF was drawn out of the cisterna magna via a 27-gauge needle attached to a catheter/syringe system. The animal was then quickly decapitated and the brain was removed. The ipsilateral parietal cerebral cortex overlying the site of contusion and ipsilateral hippocampus were quickly dissected from the brain. Contralateral samples were obtained, but not analyzed in this study. All samples were immediately frozen in liquid nitrogen and were kept frozen at −80°C until assay. Brain tissue was homogenized (7 v/w ratio) for 1 minute in a homogenization buffer of 10 mM HEPES-KOH, pH 7.9 buffer containing 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1.0 mM AEBSF, 1 :g/mL leupeptin, 1.25 :g/mL pepstatin, and 0.1% Igeral. Each sample was sonicated for 15 seconds. Homogenates were centrifuged at 12,000 g for 10 minutes at 4°C. The resulting supernatant was removed and IL-6 levels assessed in duplicate, 50 μL aliquots, using a specific ELISA (R&D Systems), according to the manufacturer's instructions. The protein level was determined by the Bradford (1976) method using reagents from Bio-Rad (Hercules, CA). Aliquots for total protein concentrations were analyzed using spectrophotometric assay at 540 nm in mg/mL based on a standard curve established for bovine serum albumin.
Statistical analysis
Data were expressed as mean values±SEM. Between-group comparisons of IL-6 data and the physiological variables were conducted using one-way ANOVA followed by multiple comparison procedures (the Tukey's Test) where appropriate. Differences were considered significant at p<0.05.
Results
Physiological parameters
Physiological parameters were obtained for each group at 3, 6, or 24 hours after TBI/normoxia, TBI/hypoxia, or sham surgery (Table 1). No animals were excluded due to mortality. Baseline physiological variables before TBI were within normal ranges. No significant difference between groups was observed in body temperature, brain temperature, or pCO2. As expected, TBI accompanied by the hypoxic insult resulted in a significant (p<0.05) decrease in the pO2 levels and MABP during the hypoxic period compared to sham or TBI-Normoxia (Bramlett et al., 1999a). For example, MABP in the 6-hour TBI-Nomoxia group at 10 minutes post-TBI was 116.12±1.71 compared to TBI-Hypoxia with a MABP of 82.20±10.23. The TBI hypoxic group also showed significant (p<0.05) reductions in pO2 values. TBI-Hypoxia animals at 30 minutes postsacrificed at 6 hours had a pO2 level of 35.35±2.41 compared to TBI-Nomoxia at 123.70±3.25. Following the hypoxic insult, TBI-Hypoxia animals returned to preinjury levels for MABP and pO2.
Values are expressed as mean±SEM.
p<0.05 versus Sham.
p<0.05 versus TBI-NO.
MABP, mean arterial blood pressure; TBI, traumatic brain injury.
Temporal profile of IL-6 protein levels
IL-6 was detected at very low levels in the sham-operated rat cortex, hippocampus, plasma, and CSF. Sham animals for each structure were compared across all time points and were not significantly different from each other (p>0.05). Therefore, these control groups were collapsed into one sham group for comparison to the temporal profile of both TBI groups. Analysis of group data at each time point showed an overall significant effect between groups in the ipsilateral cortex (p<0.007) at 3, 6, and 24 hours and hippocampus (p<0.05) at 6 hours. Post hoc multiple comparisons within the cerebral cortex showed significant (p<0.05) differences between both TBI groups at 3 and 6 hours postinjury compared to sham values (Fig. 1A). The TBI-Hypoxia group was significantly (p<0.05) different from sham at the 24-hour time point. Cerebral cortex peak values for IL-6 protein levels after TBI with or without hypoxia were present at 6 hours post-trauma. Hippocampal IL-6 data (Fig. 1B) were significantly (p<0.05) different between sham and TBI-Nomoxia at 6 hours postinjury. Therefore, within both cortical and subcortical brain structures, peak IL-6 values were present at 6 hours. CSF samples were overall significantly (p<0.05) different between groups (Fig. 1C) at 3 and 6 hours postinjury. Post hoc analysis only showed significant (p<0.05) group differences at 6 hours between TBI-Normoxia and sham. There was a moderate variability of recorded IL-6 CSF values that was probably due to the inability to obtain enough CSF from single animals for analysis and resulted in a large variation in the data within groups. Compared to brain and CSF samples, the lowest levels of IL-6 were observed in plasma after TBI. Overall analysis for the plasma levels was not significantly different between groups. However, Figure 1D clearly shows a small sustained elevation in IL-6 protein levels within the TBI-hypoxia group over time compared to TBI-normoxia and sham control animals although not significant.
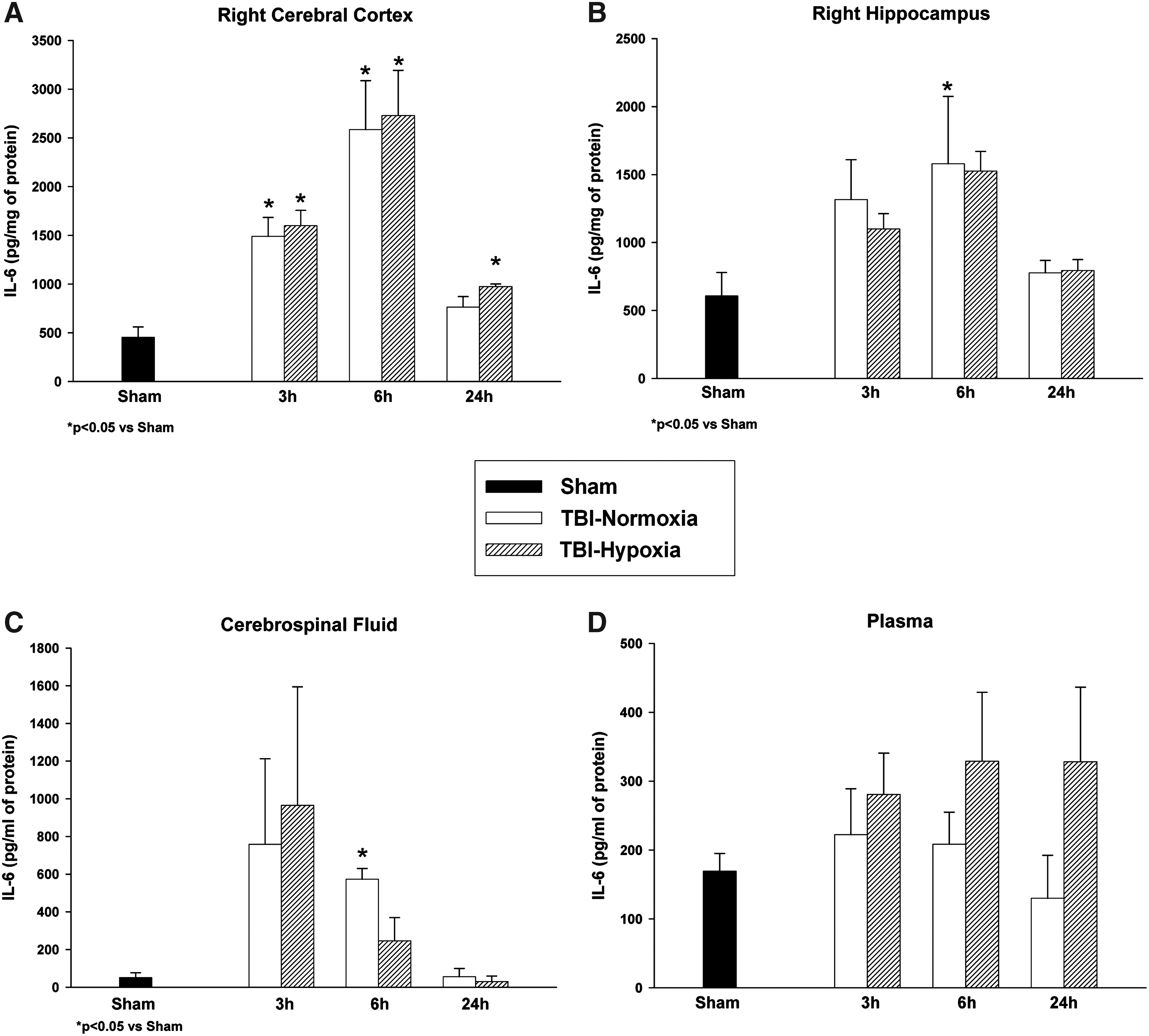
Temporal profile of interleukin (IL)-6 in the ipsilateral cerebral cortex, hippocampus, cerebrospinal fluid (CSF), and plasma samples. Following trauma, IL-6 levels were significantly increased at various times in the cerebral cortex and hippocampus
Discussion
In the present study, we report that moderate F-P brain injury leads to early and transient elevations in IL-6 levels in vulnerable brain regions, plasma, and CSF samples. At 24 hours after TBI, IL-6 values had returned to the sham-operated control levels in the cerebral cortex and hippocampus. Adding a secondary hypoxic insult to the trauma altered the IL-6 response and led to small elevations in the cerebral cortex and plasma samples. This finding may be important because secondary hypoxia in the present TBI model worsens both the histopathological and behavioral outcome (Bramlett et al., 1999a, 1999b). Thus, IL-6 may be a marker for injury severity and/or secondary insults under these experimental conditions, but due to the nonsignificant elevations, these results warrant further investigation.
IL-6 may be produced by several different cell types after TBI or other types of acute injury (Frei et al., 1989; Woodroofe et al., 1991; Taupin et al., 1993; Clark et al., 1996; Hans et al., 1999; Reyes et al., 1999; Morganti-Kossmann et al., 2007; Suzuki et al., 2009; Lu et al., 2009; Yan et al., 2012). The evidence for local synthesis of IL-6 and other cytokines by resident cells, including neurons, glia, and endothelial cells, has been reported (Ott et al., 1994; Shohami et al., 1994; Morganti-Kossmann et al., 1997; Feuerstein et al., 1998; Ghirnikar et al., 1998; Wang et al., 2001).
Brain trauma leads to a complex inflammatory response that includes microglial activation and the recruitment of circulating leukocytes (Rothwell and Strijbos, 1995; Shohami et al., 1999; Morganti-Kossmann et al., 2002). Patterns of neutrophil and macrophage accumulation after TBI have been described using a variety of animal models of brain injury (Woodroofe et al., 1991; Taupin et al., 1993; Clark et al., 1996; Chatzipanteli et al., 2000). Neutrophils and microglia have the capacity to synthesize IL-6 and may also be a source of IL-6 (Woodroofe et al., 1991; Terebuh et al., 1992). In regard to the present findings with secondary hypoxia, Bramlett et al. (1999b) reported that this complicated TBI model leads to a larger cortical contusion and aggravated hippocampal damage associated with a more robust inflammatory response. In previous studies, structural damage and an aggravated inflammatory response has been correlated with elevated IL-6 levels after brain injury (Howell et al., 2006; Yan et al., 2012). After stroke, for example, elevated levels of IL-6 are associated with the infarct size and poor outcome (Vila et al., 2000; Smith et al., 2004; Waje-Andreassen et al., 2005). In this regard, IL-6 levels are considered a highly specific biomarker of TBI severity and long-term outcome (van Griensven et al., 2003; Howell et al., 2006). In addition, IL-6 plays an important role in activating and recruiting immune cells and has been reported to suppress TNF-α and IL-1-β production and stimulate nerve growth factor (NGF) production (Kushima et al., 1992; Juttler al., 2002). In the present study, the small elevations of cortical and plasma levels of IL-6 observed in the traumatized animals undergoing a secondary hypoxic insult may play a role in the aggravated immunological–inflammatory status of the injured brain as well as a direct consequence of the increased structural damage produced by this complicated TBI model. However, further studies are warranted as these data were not as robust as seen clinically or expected in the present study.
As previously discussed, IL-6 has a multitude of actions after brain injury ranging from neuroprotective (Nijsten et al., 1987; Maeda et al., 1994; Carlson et al., 1999) to neurotoxic (Campbell et al., 1993; Chiang et al., 1994; Tilg et al., 1997; Kossmann et al., 1997; Loddick et al., 1998; Penkowa et al., 1999, 2000, 2003). In this regard, the duality of the inflammatory response to TBI in terms of potential therapeutic targets has been previously recognized (Lenzlinger et al., 2001; Morganti-Kossmann et al., 2007; Suzuki et al., 2009). While IL-6 has been reported to stimulate NGF production by astrocytes (Kossmann et al., 1996), and post-traumatic tissue repair (Swartz et al., 2001), other studies have reported that IL-6 aggravates blood–brain barrier (BBB) function (de Vries et al., 1996). Recently, transgenic mouse models of TBI have been used to investigate the importance of IL-6 on various outcome measures (Campbell et al., 1993; Stahel et al., 2000; Ley et al., 2011). Campbell et al. (1993) reported that the overexpression of IL-6, for example, was associated with sustained gliosis and a neuronal cell loss in transgenic mice. In another study by Stahel and et al. (2000), IL-6 knockout mice subjected to closed head injury demonstrated a higher mortality than their wild-type littermates, but no difference in BBB dysfunction, number of infiltrating PMNLs, or neurological outcome scores. IL-6 knockout mice recently reported by Ley et al. (2011) demonstrated poor behavioral performances and elevated IL-1β and HSP70 compared to wild-type mice. Also reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of IL-6 has also been reported (Vallieres et al., 2002). Taken together, these experimental findings support the hypothesis of dual time-dependent actions of IL-6 in the pathogenesis of brain injury (Shohami et al., 1999; Stahel et al., 2000; Morganti-Kossmann et al., 2002). Indeed, whether IL-6 and other cytokines play a protective or detrimental role after injury is dependent upon multiple factors, including the animal model being investigated, injury severity, and time-dependent cellular interactions.
Body temperature is regulated by cytokines and IL-6, a potent endogenous pyrogen, is a major regulator of the acute phase reaction and governs fever, anorexia, and apathy (Chai et al., 1996; Bluthe et al., 2000; Herrmann et al., 2003). Intraventricular administration of IL-6 is reported to elevate body temperature and reduce locomotive activity (Schobitz et al., 1993). Recently, IL-6-deficient mice following cerebral ischemia have been shown to develop profound hypothermia that was not observed in wild-type littermates (Herrmann et al., 2003). Postischemic and traumatic temperature is known to play a significant role in histopathological and behavioral outcomes (Dietrich et al., 1996; Corbett and Thornhill, 2000).
In reference to this discussion, previous clinical studies have evaluated the consequences of adult and pediatric TBI on CSF cytokine levels, including IL-6 as an indicator of functional outcome (Bell et al., 1997, 1999; Singhal et al., 2002; Chiaretti et al., 2005). Chiaretti et al. (2005) reported that increased levels of IL-6 were associated with a poor outcome, whereas other studies indicated that IL-6 levels had no association with the outcome (Bell et al., 1997, 1999; Buttram et al., 2007). Additionally, Singhal et al. (2002) concluded that peak levels of IL-6 were associated with a favorable outcome in adults. Interestingly, in the study by Buttram et al. (2007), where increased levels of IL-6 were highly variable, hypothermic treatment did not significantly attenuate the response seen in normothermic pediatric patients. In the present study, we also demonstrated a high variability in our IL-6 levels measured acutely after moderate TBI with or without hypoxia. This noted variability may be due to several factors, including variable responses to secondary hypoxia or other undefined biochemical responses to the present complicated model of TBI.
While mild reductions in temperature are protective, mild elevations worsen the outcome (Dietrich and Bramlett, 2007). In this study, we chose to maintain body temperature at normothermic levels to first document the changes in IL-6 protein levels between the simple and complicated TBI models. Based on these findings, it would be important in future studies to monitor spontaneous systemic and brain temperature alterations without a strict temperature regulation to determine whether prolonged elevations in cortical IL-6 levels correlate with increased brain temperature in the present injury model.
In this study, IL-6 levels were higher in the CSF samples, compared to those obtained from plasma. This finding is consistent with previous experimental and clinical studies that have shown higher IL-6 CSF levels compared to those reported in serum (Kossmann et al., 1995; Hans et al., 1999). Interestingly, levels of IL-6 in the CSF have been correlated with concentrations of S-100β and neuron-specific enolase in TBI patients (Pleines et al., 2001). These biomarkers are considered to be sensitive indicators of brain damage (Kim et al., 1996; Morganti-Kossmann et al., 2002). In our study, CSF levels of IL-6 were elevated early after trauma, had returned to normal levels by 24 hours, and did not differ significantly between normoxic and hypoxic TBI rats. It may be important in future studies to experimentally assess the relationships between maximum levels of IL-6 in CSF samples and trauma severity. It should also be noted that our model of secondary hypoxia is not associated with hypercapnia, a common event associated with hypoxia.
In summary, the present results show that experimentally induced F-P brain injury induces significant elevations in IL-6 protein levels in vulnerable brain regions, including the cerebral cortex and hippocampus. Within the cerebral cortex, secondary hypoxia, which is known to aggravate the histopathological and behavioral outcomes after TBI produced prolonged elevations in the IL-6 protein that were significantly different from sham-operated controls. Acute elevations in IL-6 were also seen in plasma and CSF samples. Future studies will be aimed at clarifying the role of IL-6 in the detrimental effects of secondary hypoxia following TBI and the importance of this cytokine as a target for early therapeutic interventions (Beauchamp et al., 2008; Lu et al., 2009; Clausen et al., 2011).
Footnotes
Acknowledgments
This study was supported by NIH/NINDS grants P50NS30291 and RO1NS42133 and Veterans Affairs 1 I01 BX000521. The authors would like to thank Jeremy Lytle for editorial assistance and manuscript preparation.
Disclosure Statement
No competing financial interests exist.