
Abstract
Reperfusion therapies for stroke diminish in effectiveness and safety as time to treatment increases. Hypothermia neuroprotection for stroke is established, but its clinical translation has been hampered by uncertainties regarding optimal temperature and complications associated with moderate hypothermia. Also, hypothermia targeting temperatures of 32–33°C is associated with clinical and logistical problems related to induction and adverse side effects. We hypothesized that ischemic damage and tPA-exacerbated blood/brain barrier (BBB) breakdown produced following 30 minutes of middle cerebral artery occlusion and either 1 hour of saline or tPA infusion would be reduced by treatment with very mild cooling of 1.5°C for 48 hours followed by 24 hours of gradual rewarming. Infarct volume was reduced by 29.6% (p<0.001) and 41.9% (p<0.001) in hypothermic-tPA (Hypo_tPA)-treated and hypothermic-saline (Hypo_Sal)-treated animals compared to normothermic-tPA (Norm_tPA) and saline (Norm_Sal)-treated animals, respectively. Hypothermia also reduced IgG extravasation in tPA-treated, but not saline-treated groups compared to their normothermic controls (p<0.001). The ipsilateral–contralateral changes in optical density for IgG extravasation were 18.4% greater in the Norm_tPA than Norm_Sal (p<0.001) group. The ipsilateral–contralateral changes in optical density for IgG extravasation were reduced by 17.8% (p<0.001) in the Hypo_tPA compared to Norm_tPA group. No significant mean difference in IgG extravasation was seen between Hypo_tPA and Hypo_Sal groups (p>0.05). Very modest hypothermia to reduce the BBB breakdown could improve the availability and safety of reperfusion treatments for stroke.
Introduction
T
There are multiple potential reasons for reduced response or poor outcomes to reperfusion therapies following stroke, but it is likely that key factors include the severity of the initial ischemic injury and exacerbating secondary injury processes causing increased blood/brain barrier (BBB) breakdown that occurs following reperfusion (Yenari et al., 1995; Wilde, 1997; van der Pals et al., 2011).
Moderate therapeutic hypothermia has been shown to be a robust neuroprotectant in both preclinical stroke studies and human cardiac arrest, where the target core temperature has been below 34°C (Bernard et al., 2002; van der Worp et al., 2007; Jacobs et al., 2013). Hypothermia may also offer the opportunity to prolong the therapeutic window for the implementation of acute stroke therapies, reduce stroke injury related to ischemia–reperfusion, and improve safety. However, such moderate hypothermia creates its own practical problems specifically related to the logistics of inducing hypothermia and its complications, including sepsis and cardiac/hemodynamic instability (Hemmen and Lyden, 2007).
There are additional potential limitations of using moderate hypothermia (defined here as a target temperature of 32–33°C) in that, it can potentially induce a hypercoagulable state, impair platelet function, and can be associated with an increased risk of hemorrhagic transformation (Wilde, 1997; Schwab et al., 2001). Despite decades of research using hypothermia for neuroprotection, fundamental knowledge for optimal depth and duration remains elusive.
One potential way in which hypothermia could act to improve stroke outcome is by its robust ability to protect the BBB following ischemic stroke (Smith and Hall, 1996; Chi et al., 2001; Hammer and Krieger, 2003). Recently, Tang et al. (2013) showed that brief (3 hours) moderate hypothermia (33°C) postischemia could ameliorate BBB disruption and tPA-related hemorrhage. In addition, prolonging cooling duration may further improve outcomes (Corbett et al., 2000).
In this study, we propose that prolonged (48 hours) very mild therapeutic hypothermia (defined here as 1.5°C reduction on core temperature) has the capability of mitigating stroke severity and tPA-induced BBB breakdown during ischemia–reperfusion. We hypothesized that very mild hypothermia following 30 minutes of middle cerebral artery occlusion (MCAO) in the mouse would reduce infarct volume and BBB breakdown compared to normothermic (36.5°C) control with or without tPA treatment. Although targeting mild hypothermia has been shown to reduce infarct volume, no study to our knowledge has looked at the benefit of prolonged mild cooling in conjunction with tPA treatment. Demonstrating the effectiveness of a very mild but rather prolonged temperature reduction irrespective of tPA treatment would facilitate translation of hypothermia treatment regimens to human stroke (Kammersgaard et al., 2000).
Materials and Methods
Sixty animals were used in five experimental groups. All experiments and procedures were approved by the local animal care committee and were in accordance with the Canadian Council on Animal Care guidelines. Six male C57 black mice (3 months old, 25–35 g; Charles River Breeding Laboratories) were prepared for 30 minutes of transient MCAO using the intraluminal filament method and were randomly assigned before the surgical preparation to the two active treatments performed unblinded (1 hour tPa or saline infusion and mild hypothermia at 35.0°C for 48 hours or normothermia at 36.5°C). Temperature for all animals was controlled at 36.5°C during the animal preparation, transient MCAO, and saline or tPA treatment, after which the randomized temperature protocol was initiated.
There were three normothermic groups (maintained at 36.5°C): Sham (sham MCAO, i.v. saline immediately postreperfusion); Norm_Sal (i.v. saline immediately postreperfusion); and Norm_tPA (i.v. tPA immediately postreperfusion). There were two mild hypothermic groups: Hypo_Sal (i.v. saline immediately postreperfusion) and Hypo_tPA (i.v. tPA immediately postreperfusion). In these mild hypothermic animals, the core temperature was lowered to 35°C for 48 hours after tPA/saline infusion was completed (1 hour after ischemia–reperfusion) followed by gradual rewarming for 24 hours.
Animals were anesthetized with 1.5–2.5% isoflurane in 30% oxygen in nitrogen. Radio transmitter probes that continuously recorded temperature (Model TA10TA-F20; DataScience, Inc.) were implanted through an incision along the abdomen, as described by Colbourne et al. (1996). The wound was sutured with 5-0 VICRYL and 4-0 silk (Ethicon). Buprenorphine (0.05 mg/kg, s.c.) provided analgesia and fluids were replenished (0.5–1 mL sterile saline I.P., Braun Medical, Inc.). Mice weighed 27–34 (29.17±1.98) g, inclusive of radio probes.
Mice recovered from transmitter implantation at least 7 days before transient MCAO was performed, as described previously (Barber et al., 2004). Transcranial measurements of relative cerebral blood flow (CBF) were made by laser-Doppler flowmetry. A 0.5-mm-diameter microfiber laser-Doppler probe (Probe 418; Perimed) was attached to the skull with cyanoacrylate glue 6 mm lateral and 1 mm posterior of bregma. The severity of the reduction of rCBF during MCAO was predefined (i.e., <70% for animals to be included).
The dose of tPA (10 mg/kg, Activase® compared to 0.9 mg/kg used clinically) was selected based on a 10-fold higher rate of metabolism of tPA in the rodents compared to humans (Korninger and Collen, 1981). After 30 minutes of MCAO, 10% of the saline or tPA dose was administered i.v. as a bolus, with the remaining 90% given over 1 hour through the left tail vein using a syringe held infusion pump (Harvard Apparatus 11 plus). Buprenorphine (0.05 mg/kg, s.c.) analgesia was administered before recovery from anesthesia.
Normothermia was maintained at 36.5°C for 72 hours. Hypothermia was initiated at the end of tPA/saline treatment (1 hour postreperfusion) and maintained at 35°C for 48 hours followed by gradual rewarming. Temperature was monitored for 3 days in freely moving animals housed in a day/night light cycle. Thermoregulation and temperature data were managed by the DataQuest Thermoreg (DataSciences, Inc.) software and acquisition programs (Jacobs et al., 2013). Abdominal temperature data were sampled at 30-second intervals. Heating was provided by a 90W heat lamp and cooling by a 110W fan situated 50 cm directly above the animal's recovery housing.
All animals were euthanized at 72 hours and brains were processed for histology. Animals were deeply anesthetized with sodium pentobarbital (70 mg/kg intraperitoneally) and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde. The brain was then embedded in paraffin and sectioned at 10 μm thickness.
Histology was assessed in sections blinded to treatment groups. Hematoxylin and eosin (H&E)-stained sections were used to determine infarct volumes as a % of the hemisphere corrected for edema, as described previously (Kaur et al., 2011). Infarct volume was calculated from a minimum of seven slices through the cerebrum, with a distance between sections of approximately 1 mm. BBB breakdown was determined by systemic IgG extravasation into the parenchyma assessed in coronal sections (Bregma: 1.2 mm) stained with the goat anti-rat IgG antibody conjugated to horseradish peroxidase. Diaminobenzidine was used for visualization of the antibody reactivity.
An Olympus BX 61 (Olympus Canada) light microscope at ×1.5 magnification was used to quantify IgG extravasation by measuring the gray scale value using comparative regions of interest in both cerebral hemispheres and subtracting the gray scale intensity (in arbitrary units) of the contralateral and ipsilateral stroke regions. The results are presented as a ratio of contralateral to ipsilateral difference of optical density (inverse gray scale values). With increasing BBB injury, a darker staining (increased optical density) was observed, indicative of greater extravasation of IgG (mol weight ∼70 kDa) (Kaur et al., 2011).
After animals for sham surgery were identified (N=6), animals were randomly allocated to four treatment groups as stated above (N=54). The sample size for each of these groups was a minimum of 13 animals and included animal losses related to attrition (e.g., surgical complications, morbidity, and increased bleeding) and protocol violations (e.g., failure to meet thermoregulatory target temperature) to demonstrate differences of 20% between groups considering an SD of 10%, a power of 0.8, and an alpha of 0.05. All data are reported as mean±SD. Mean differences between groups were analyzed by 2-way ANOVA applying a Bonferroni correction. Differences were considered statistically significant at p<0.05.
Results
Forty of the 60 mice were included in the analysis. In total, 11 (19%) of the animals reached their humane endpoints before the 72-hour euthanasia mark. Seven of these animals died due to intraoperative surgical complications, before the thermoregulation protocol started. Four animals died postoperatively (Norm_tPA=1, Hypo_sal=2, Hypo_tPA=1), each due to postsurgical complications and unrelated to the treatment group. Nine additional mice did not meet thermoregulatory criteria (±0.5°C from target temperature): 2 Norm_tPA, 5 Hypo_Sal, and 2 Hypo_tPA. One animal had inadequate H&E (N=39) staining for analysis, and two animals had inadequate IgG histological sections (N=38). The mean relative CBF decreases during sham or MCAO were as follows: Sham 8.6%±17 (n=6), Norm_Sal 79.1%±18 (n=5), Norm_tPA 83.6%±8.0 (n=10), Hypo_Sal 76.2%±11 (n=6), and Hypo_tPA 72.0%±8.0 (n=13).
There was no statistical difference between MCAO groups (p>0.05 1-way ANOVA). The CBF change in the sham occlusion group was significantly less than that in all MCAO groups (p<0.001 1-way ANOVA). Immediately during reperfusion, the CBF measure returned to at least 75% of the baseline preocclusion value; there was no statistical difference between the treatment groups.
Thermoregulatory data are shown in Figure 1. The mean intraischemic temperature was 36.2°C±0.8°C for all groups with no significant difference in the intraischemic temperature between groups (p>0.05 1-way ANOVA). The postischemic temperatures for the first 48 hours before rewarming in the hypothermia groups were as follows: Sham 36.45°C±0.53°C, Norm_Sal 36.4°C±0.32°C, Norm_tPA 36.4°C±0.45°C, Hypo_Sal 35.08°C±0.5°C, and Hypo_tPA 35.04°C±0.35°C. Temperature control during this period was statistically different for hypothermic groups when compared to normothermic groups (p<0.001, 1-way ANOVA), but not between hypothermic groups (p>0.05, 1-way ANOVA) nor between the three normothermic groups (p>0.05, 2-way ANOVA).
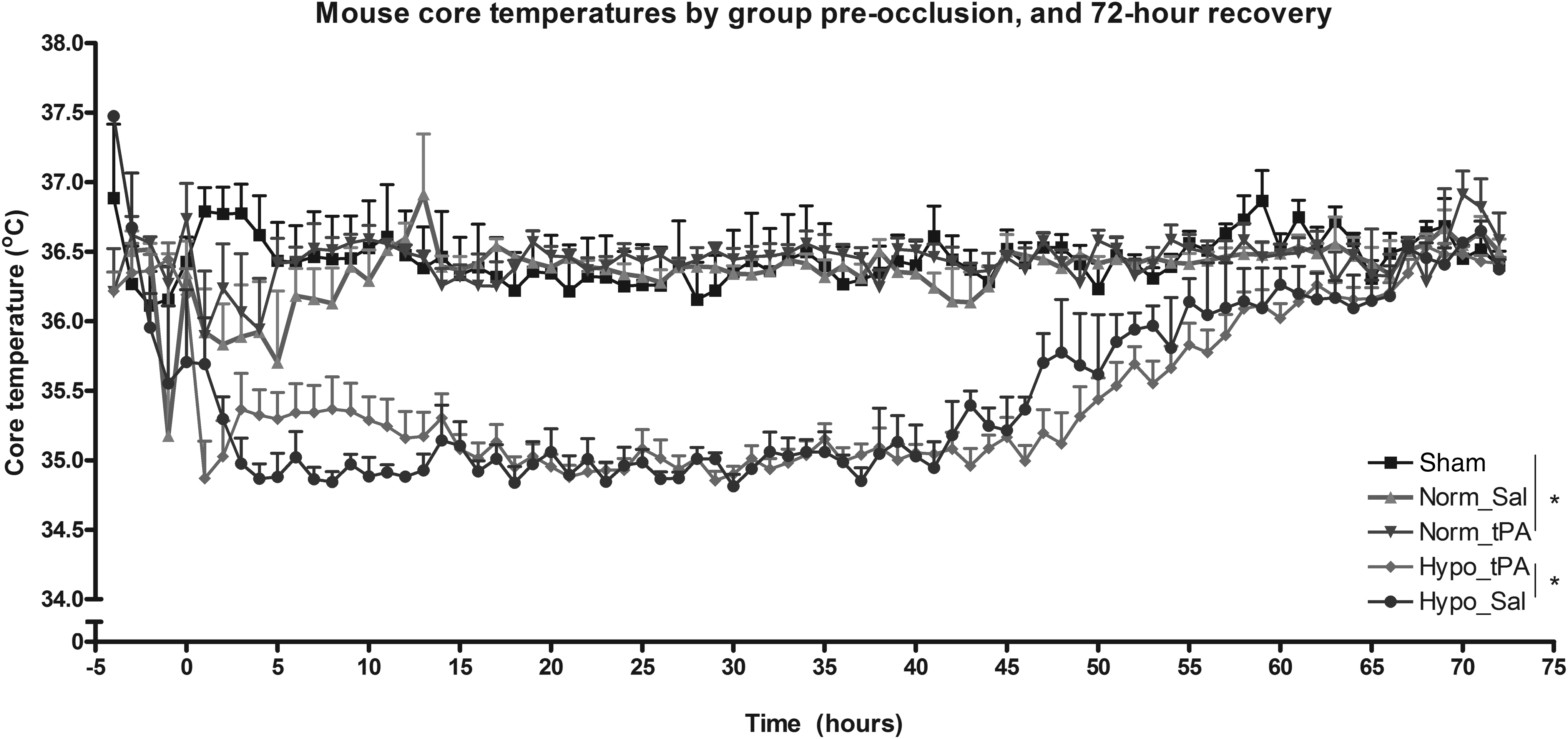
Mouse core temperatures sampled twice each minute and presented as 60-minute averages for the normothermic (Norm_Sal/tPA) and hypothermic groups (Hypo_Sal/tPA) at the onset of reperfusion. Target temperature was maintained for over 48 hours and was significantly less in hypothermic than normothermic groups (*p<0.001).
Assessment of hemispheric brain infarct volume is shown in Figure 2. IgG extravasation, determined by the optical density value ratio between the contralateral and ipsilateral, is shown in Figure 3. In the Hypo_Sal group, hypothermia significantly reduced mean percentage hemispheric infarct volume (6.4%±10.24) compared to the Norm_Sal group (35.7%,±8.77, p<0.01). Similarly, in Hypo_tPA, hypothermia significantly reduced mean percentage hemispheric infarct volume (18.7%,±11.53) compared to the Norm_tPA group (48.3%,±14.5, p<0.001). tPA treatment was not associated with an increase in infarct volume when compared with normothermic saline-treated animals (p>0.05). No evidence of hemorrhagic transformation was observed.
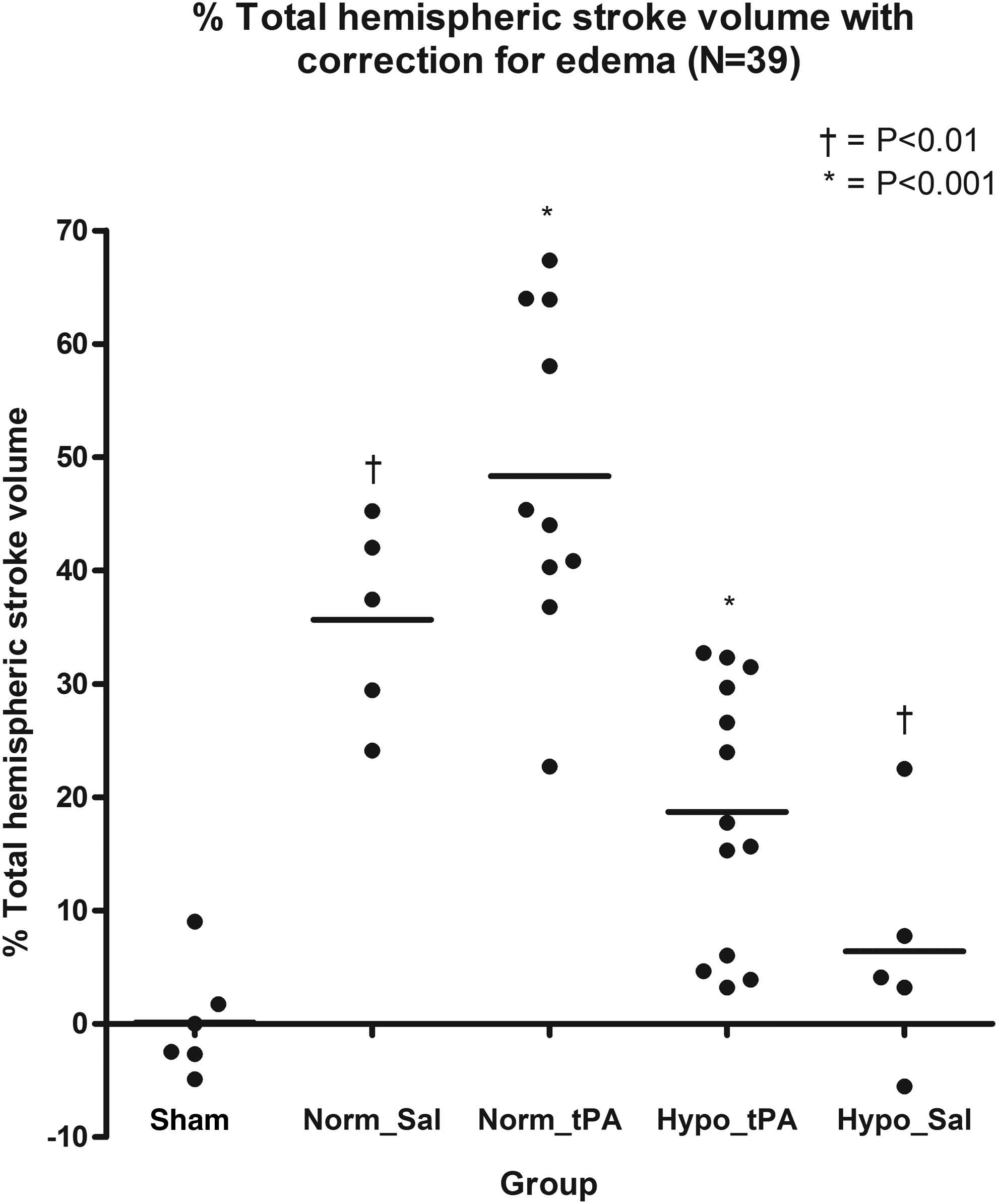
Volume of cortical infarction in mice subjected to transient middle cerebral artery occlusion and treated with saline (Norm_Sal) or tPA (Norm_tPA) immediately following reperfusion. Hypo_Sal and Hypo_tPA animals had significantly reduced infarct volume from Norm_Sal (*p<0.001) and Hypo_tPA significantly different from Norm_tPA (†p<0.01).

Evidence for reduced BBB dysfunction with hypothermia treatment. Contralateral–ipsilateral differences in staining for IgG as a percentage of that contralaterally within the infarct region in Norm_Sal, Norm_tPA, Hypo_Sal, and Hypo_tPA groups. IgG staining was significantly less in tPA-treated animals that were hypothermic during reperfusion. Also, IgG extravasation was increased in tPA-treated groups compared to Norm_Sal and Sham groups. Furthermore, mild hypothermia modified the effect of tPA-increased IgG extravasation. Norm_tPA significantly different from all other groups (*p<0.001).
Increased optical density confirmed BBB breakdown and IgG protein extravasation in the ischemic hemisphere in all animal groups. The ipsilateral–contralateral changes in optical density for IgG extravasation were 18.4% greater in the Norm_tPA than Norm_Sal (p<0.001) group. The ipsilateral–contralateral changes in optical density for IgG extravasation were reduced by 17.8% (p<0.001) in Hypo_tPA compared to Norm_tPA. No significant mean difference in IgG extravasation was seen between the Hypo_tPA and Hypo_Sal groups (difference = −0.84%, p>0.05).
Discussion
The novel finding in this study was that prolonged mild cooling (1.5°C) for 48 hours attenuates tPA-exacerbated BBB breakdown and reduced infarct volume following ischemia–reperfusion. tPA administration was associated with increased BBB breakdown, but not an increase in infarct volume. Prolonged mild hypothermia was associated with reduced infarct volume in both tPA-treated and saline-treated animals, and reduced IgG extravasation was confirmed only in tPA-treated mice. This supports the concept that very mild therapeutic hypothermia could be an important adjuvant therapy to reperfusion strategies by reducing the BBB breakdown and lengthening the duration of the therapeutic time window.
Unlike what is commonly seen in human stroke treatment, we did not observe hemorrhagic transformation in this model. This may be related to several factors, including stroke severity, timing of treatment and recanalization, age of animal, and cooling parameters. Kaur et al. (2011) only observed hemorrhagic transformation in elderly rats treated with tPA, but not young healthy animals.
While previous studies have demonstrated that moderate hypothermia reduces both infarct volume and blood/brain barrier breakdown, the benefit of very mild hypothermia has not been consistently demonstrated (Kallmunzer et al., 2012). This likely relates to differences in severity of the stroke model, the time of hypothermia induction, and the duration of hypothermia (Kollmar et al., 2007; Lakhan and Pamplona, 2012; Campbell et al., 2013). Mild hypothermia in our study was most effective in animals that received tPA, and previous studies have shown that hypothermia maintains microvascular integrity and reduces hemorrhages, possibly mediated by attenuating the activities of MMP2, MMP9, UPA, and tPA (Hamann et al., 2004; Barber et al., 2012).
In our study, mild hypothermia was induced after administration of tPA. Earlier induction of hypothermia might have produced a more profound response on ischemic injury, but we considered the delayed induction of cooling to be more clinically relevant.
Our findings may have wider implications for the extension of the time window for administration of tPA and the safety of its administration under conditions of prolonged mild hypothermia. There have been concerns of therapeutic hypothermia reducing the effectiveness of tPA, but the effect of even moderate hypothermia on the fibrinolytic activity of tPA is likely modest (Yenari et al., 1995; Shaw et al., 2007). However, moderate hypothermia can contribute to significant hematological side effects such as hypercoagulable state, thrombocytopenia, and hemorrhagic transformation (Schwab et al., 2001); each of these hematological responses to hypothermia is related to the severity of temperature reduction. Therefore, milder hypothermia may have advantages in reducing the hematological consequences observed with moderate therapeutic hypothermia.
Recent clinical studies have shown the feasibility of performing mild 2.5°C hypothermia (to 35°C) with cold saline and surface cooling, and MR imaging studies have demonstrated reduced cerebral edema associated with such mild hypothermia (Bi et al., 2011; Hong et al., 2014; Piironen et al., 2014). Future studies would need to be performed to confirm the relative effectiveness of milder hypothermia compared with deeper levels of therapeutic hypothermia regarding their effects on reducing stroke volume and BBB breakdown following tPA administration, specifically utilizing an animal clot model of MCAO.
Our study has several limitations. We used a relatively short survival time of 3 days poststroke. Examining histology after brief recovery has limitations with respect to delayed cellular death occurring over days and weeks. Maintaining core temperature within±0.5°C was challenging in our system, specifically targeting a small cooling target temperature of 1.5°C, which is reflected in the seven hypothermic mice not meeting criteria versus two normothermic-treated animals. Postoperative mortality was due to surgical complications and was not significantly different between hypothermic and normothermic groups (Fisher's exact test p=0.63), consistent with the findings of Tang et al. (2013) that hypothermia was not associated with increased mortality.
We did not elect to perform behavioral studies because of the single practical reason that mice were undergoing hypothermia close to the euthanasia time. As we did not compare diverse periods of hypothermia in our model, it is unknown whether the 48-hour period of hypothermia was necessary or sufficient and whether this therapy would universally be maximally efficacious.
In summary, our results have shown that a very mild but prolonged hypothermia can reduce both BBB breakdown and infarct volume following ischemia–reperfusion. We observed that the therapeutic effect of hypothermia on the BBB is most profound in animals that have been administered tPA. The potential clinical implications of this study include a simplification of hypothermia induction protocols and improved generalizability of therapeutic hypothermia. Beyond the results of this study, the next essential step in therapeutic advancement for stroke will be to demonstrate the mitigation of ischemic and reperfusion-related brain damage following reperfusion therapies by exceptionally mild hypothermia protocols.
Footnotes
Acknowledgments
This project was supported by the Canadian Institute of Health Research. Undergraduate studentship funding was provided by The O'Brien Centre (University of Calgary, Canada) and the Markin USRP (University of Calgary, Canada).
Author Disclosure Statement
No competing financial interests exist.