
Abstract
This study aimed to determine a target temperature for intraischemic hypothermia that can block increases in extracellular glutamate levels. Two groups of 10 rats each formed the normothermia and intraischemic hypothermia groups. Extracellular glutamate levels, the extracellular potential, and the cerebral blood flow were measured at the adjacent site in the right parietal cerebral cortex. Cerebral ischemia was induced by occlusion of the bilateral common carotid arteries and hypotension. In the intraischemic hypothermia group, brain hypothermia was initiated immediately after the onset of membrane potential loss. In the normothermia group, extracellular glutamate levels began to increase simultaneously with the onset of membrane potential loss and reached a maximum level of 341.8 ± 153.1 μmol·L−1. A decrease in extracellular glutamate levels was observed simultaneously with the onset of membrane potential recovery. In the intraischemic hypothermia group, extracellular glutamate levels initially began to increase, similarly to those in the normothermia group, but subsequently plateaued at 140.5 ± 105.4 μmol·L−1, when the brain temperature had decreased to <32.6°C ± 0.9°C. A decrease in extracellular glutamate levels was observed simultaneously with the onset of membrane potential recovery, similarly to the findings in the normothermia group. The rate of decrease in extracellular glutamate levels was the same in both groups (−36.6 and −36.0 μmol·L−1 in the normothermia and intraischemic hypothermia groups, respectively). In conclusion, the target temperature for blocking glutamate release during intraischemic hypothermia was found to be 32.6°C ± 0.9°C. Our results suggest that the induction of intraischemic hypothermia can maintain low glutamate levels without disrupting glutamate reuptake. Institutional protocol number: OKU-2016146.
Introduction
T
Recently, it has become possible to reduce primary injury using intranasal cooling or pharyngeal cooling without disturbing the ROSC in clinical situations because brain temperature is selectively decreased (Castren et al., 2010; Takeda et al., 2012, 2014). Furthermore, it was reported that the early induction of hypothermia during cardiac arrest improves neurological outcomes in patients who undergo extracorporeal cardiopulmonary resuscitation (ECPR) (Nagao et al., 2010). There is growing evidence that ECPR with hypothermia leads to a favorable neurological outcome regardless of patients being in the hospital or out of the hospital (Sakamoto et al., 2014; Tan, 2017). All of these cooling methods can cool the brain during a cardiac arrest without disturbing the ROSC. Although it has been demonstrated that brain hypothermia induced before ischemia abolishes increases in extracellular glutamate levels (Busto et al., 1989; Patel et al., 1994), no studies have reported the target temperature for intraischemic hypothermia based on dynamic changes in glutamate levels.
The present study was designed to determine the target temperature for suppressing increases in extracellular glutamate levels during the loss and recovery of membrane potential in rat models. We serially measured glutamate levels every minute and continuously measured extracellular potential and cerebral blood flow (CBF) at the adjacent site in the right parietal cortex.
Materials and Methods
A total of 20 male Sprague-Dawley rats (Charles River Japan, Yokohama, Japan) weighing 311 ± 12 g were used in this study. The sample size was estimated using a power analysis based on the results of a previous study using the same animal model (actual power, 0.85; total sample size, 9) (Mizoue et al., 2015). The animals were housed under specific pathogen-free conditions in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Animals were fasted overnight before the experiments, although they had ad libitum access to water. All experiments were performed in accordance with the National Institutes of Health Animal Care Guidelines and were approved by the Animal Research Control Committee of Okayama University Medical School, Okayama City, Japan.
General protocol
The experiment was performed in a university hospital laboratory. The animals were divided into two groups of 10 rats each: a normothermia group and an intraischemic hypothermia group. Anesthesia was induced using 4% isoflurane in oxygen. After tracheal intubation and the initiation of artificial ventilation (SN-408-7; Shinano, Tokyo, Japan), anesthesia was maintained using 1.5% isoflurane in 60% oxygen balanced with nitrogen. Polyethylene catheters were placed in the right femoral artery for continuous monitoring of mean arterial blood pressure and blood sampling, and in the right femoral vein for administration of heparin and exsanguination. After placement of the animal in a stereotaxic apparatus (Narishige, Tokyo, Japan), a borosilicate glass electrode (tip diameter <5 μm) was placed through a burr hole into the fifth layer of the right parietal cerebral cortex (3 mm posterior and 3 mm right of the bregma, 0.75 mm below the cortical surface) for monitoring the extracellular potential. Adjacent to the extracellular electrode, a microdialysis probe (membrane length, 1.0 mm; molecular weight cutoff, 50,000; outer diameter, 220 μm [A-I-4-010; Eicom, Kyoto, Japan]) was placed 1.25 mm below the cortical surface for the serial measurement of glutamate levels, and a laser Doppler flow probe (FLO-C1; Omegawave, Tokyo, Japan) was placed on the dura for continuous monitoring of regional CBF. Brain temperature was monitored using a small thermocouple placed in the left parietal epidural space and controlled by a gentle flow of warmed saline coursing over the skull surface. Rectal temperature was monitored and maintained at 37.0°C ± 0.5°C using a heated water blanket.
Ischemia method
The experimental protocol is outlined in Figure 1. After occlusion of the bilateral common carotid arteries (two-vessel occlusion [2VO]), CBF was decreased at a rate of 2.5% of the baseline level/min by draining the venous blood until a sudden negative shift in the extracellular potential (i.e., membrane potential loss) was observed. CBF was maintained at the level of membrane potential loss for 5 minutes, following which it was restored by the return of blood flow at the same rate. The beginning of membrane potential loss was defined as a sudden negative shift in the extracellular potential. The beginning of membrane potential recovery was defined as a positive shift in the extracellular potential for >1 minute.
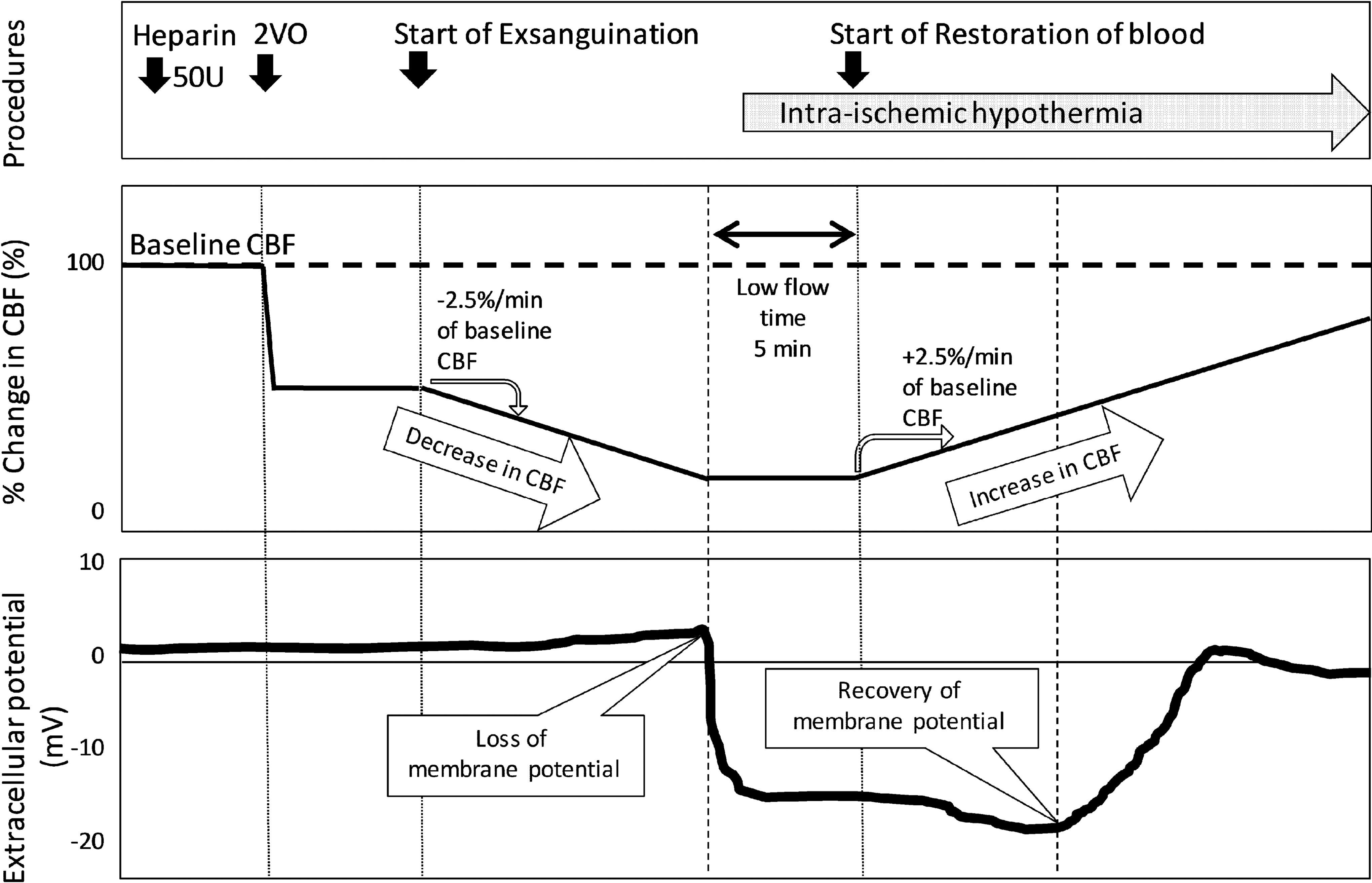
Experimental protocol. The percent change in the CBF and extracellular potential are shown. The baseline CBF values were measured after surgery. The bilateral common carotid arteries were occluded 5 minutes after heparin injection (50 U). Five minutes later, CBF was continuously decreased by exsanguination of the venous blood at a rate of 2.5% of the baseline level until a sudden negative shift in the extracellular potential (loss of membrane potential) was observed. CBF was maintained at the level of membrane potential loss for 5 minutes, following which it was increased by the return of blood flow at the same rate. The shaded arrow region indicates the duration of brain hypothermia initiated immediately after membrane potential loss. CBF, cerebral blood flow.
Measurement of glutamate levels
The microdialysis probes were perfused with Ringer's solution at a flow rate of 2 μL·min−1 using an infusion pump (ESP-32; Eicom). The dialysate was automatically collected every minute using a fraction collector (EFC-82; Eicom) in a sample tube containing 10 μL of Ringer's solution (total 12 μL for each sample) to prevent evaporation. Glutamate was quantified using an electrochemical detector (ECD) with high-performance liquid chromatography and a computerized control system (Nanospace Syscon 21; Shiseido, Tokyo, Japan). Glutamate was isolated from the samples using a separation column (4.6 × 150 mm, 4 μm, GU-GEL; Eicom) and oxidized by an immobilized enzyme column (3.0 × 4.0 mm, 37–74 μm, E-ENZYMPAC; Eicom) to hydrogen peroxide, which was detected by the ECD. The mobile phase consisted of 250 mg·L−1 cetrimonium bromide (C19H42BrN) and 0.05 mg·L−1 EDTA sodium in 60 mmol·L−1 NH4Cl-NH4OH solution.
Cooling method
Nasopharyngeal cooling (Hagioka et al., 2003) was initiated immediately after the onset of membrane potential loss. Cannulae (20-gauge) were inserted into both nasal cavities to a depth of 5 mm. Cold physiological saline (5°C) was infused at a rate of 100 mL min−1 kg−1 of body weight using a roller pump until the epidural temperature had decreased to 31°C. The cold saline flowed out of the oral cavity and was aspirated by a suction device to prevent the animal from becoming wet. This method enabled a rapid decrease in the temperature of the entire brain without a decrease in the body temperature. Hypothermia was maintained until the end of membrane potential recovery. The rectal temperature was maintained at 37.0°C ± 0.5°C during this period.
Statistical analyses
All values are expressed as means ± standard deviations. The glutamate and extracellular potential data were analyzed using repeated-measures analysis of variance followed by Fisher's post hoc test. All other statistical analyses were performed using Student's t-test (two tailed). A p-value of <0.05 was considered statistically significant in all tests.
Results
All parameters were maintained within normal ranges before the initiation of ischemia. Changes in CBF, the extracellular potential, extracellular glutamate levels, and the epidural temperature are shown in Figure 2.

Changes in the CBF
In the normothermia group, the CBF threshold for membrane potential loss was 21.2% ± 7.9% of the control level (Fig. 2A). Extracellular glutamate levels (preischemic value, 32.1 ± 34.4 μmol·L−1) began to increase simultaneously with the onset of membrane potential loss and reached a maximum level of 341.8 ± 153.1 μmol·L−1 (Fig. 2B, C). A decrease in extracellular glutamate levels was observed simultaneously with the onset of membrane potential recovery. The CBF threshold for membrane potential recovery was 44.9% ± 21.6% of the control level (Fig. 2A).
In the intraischemic hypothermia group, the CBF threshold for membrane potential loss was 20.8% ± 10.5% of the control level (p = 0.94 vs. normothermia group; Fig. 2A). Extracellular glutamate levels (preischemic value, 28.1 ± 39.8 μmol·L−1; p = 0.75) initially began to increase, similarly to those in the normothermia group, and subsequently plateaued at 140.5 ± 105.4 μmol·L−1, when the brain temperature had decreased to <32.6°C ± 0.9°C (Fig. 2B–D). The 95% confidence interval of the target temperature for intraischemic hypothermia required to block the increase in extracellular glutamate levels in rats was 32.0–33.2°C. A decrease in extracellular glutamate levels was observed simultaneously with the onset of membrane potential recovery, similarly to the findings in the normothermia group. The CBF threshold for membrane potential recovery (26.3% ± 9.3% of the control level) was significantly lower in this group than in the normothermia group (p = 0.02; Fig. 2A). Extracellular glutamate levels at this point were also significantly lower (125 ± 96 μmol·L−1) than those in the normothermia group (317 ± 155 μmol·L−1; p = 0.004; Fig. 2C).
Extracellular glutamate levels decreased from 104 ± 16 to 31 ± 12 μmol·L−1 in the normothermia group and from 112 ± 12 to 40 ± 17 μmol·L−1 in the intraischemic hypothermia group, with decrease rates of −36.6 and −36.0 μmol·L−1·min−1, respectively (Fig. 3).
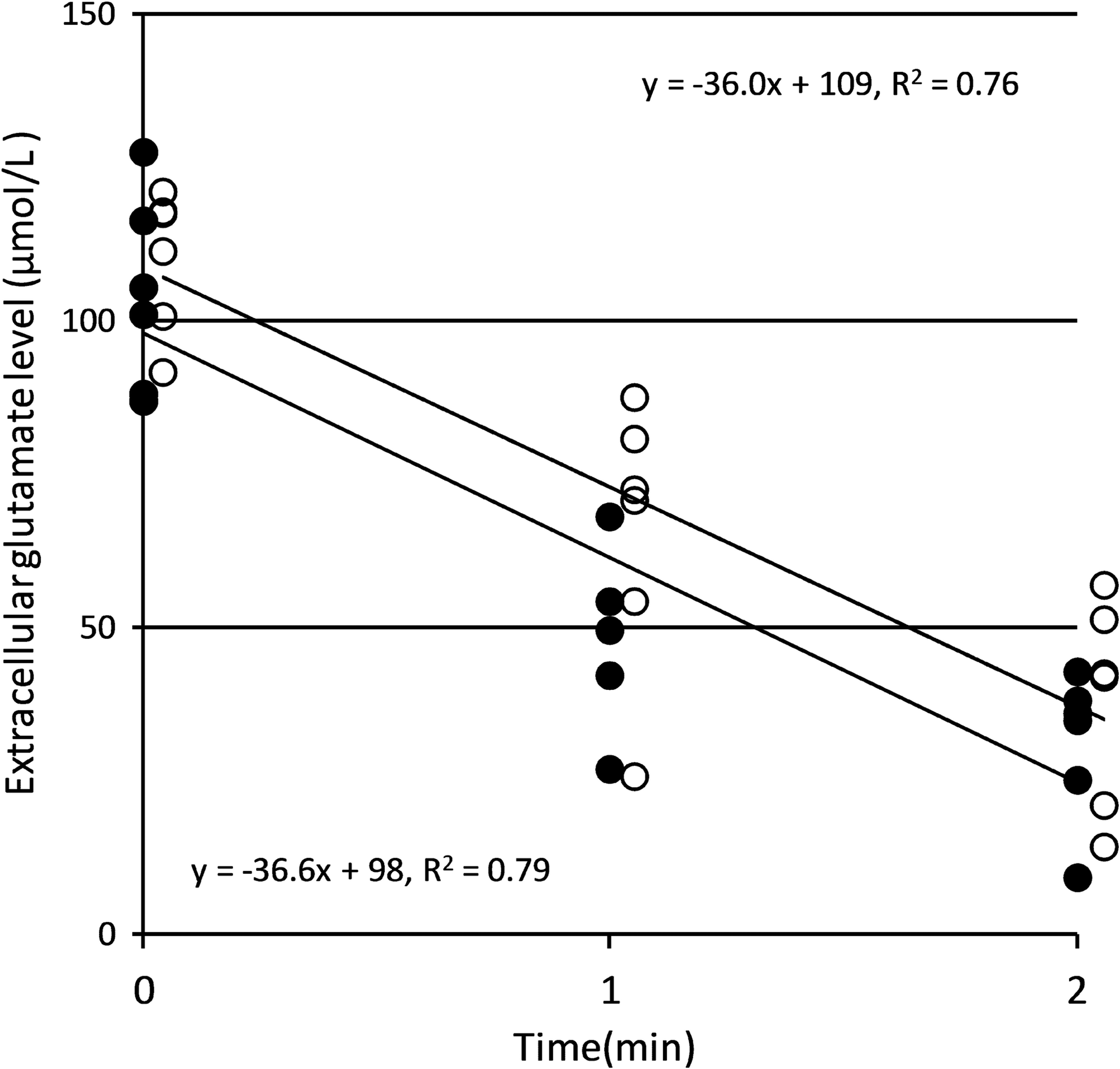
Rate of decrease in extracellular glutamate levels. The filled circles and open circles indicate the values for the normothermia and intraischemic hypothermia groups, respectively. The rates of decrease in extracellular glutamate levels, shown by the inclination of each line, are −36.6 and −36.0 μmol·L−1·min−1 in the normothermia and intraischemic hypothermia groups, respectively.
Discussion
To the best of our knowledge, the present study is the first to determine the target temperature for intraischemic hypothermia in a rat model. By decreasing the brain temperature to <32.6°C ± 0.9°C (95% confidence interval: 32.0–33.2°C), intraischemic hypothermia can block increases in extracellular glutamate levels. Table 1 summarizes the results of previous studies investigating the effects of hypothermia initiated “before ischemia” on glutamate levels. Extracellular glutamate levels did not increase when the brain temperature was <33°C, regardless of the animal species (e.g., rats and rabbits), region of the brain (e.g., striatum, dorsal hippocampus, and left parietal cortex), method of inducing brain ischemia (e.g., four-VO plus hypotension, neck tourniquet plus hypotension, and cardiac arrest), or the duration of ischemia (e.g., 5, 7.5, 15, and 20 minutes) (Globus et al., 1988; Busto et al., 1989; Illievich et al., 1994a, 1994b; Nakashima and Todd, 1996; Takata et al., 2005). These observations suggest that the mechanisms of glutamate release are common and blocked when the brain temperature is <33°C, regardless of whether hypothermia is initiated before or after the onset of ischemia.
Glutamate level is expressed relative to the preischemic value, shown as 100%. Values in parentheses indicate temperature in degrees centigrade. The increase in the extracellular glutamate level is greatly reduced when the brain temperature is less than 33°C.
4VO, four-vessel occlusion.
In the present study, intraischemic hypothermia did not decrease extracellular glutamate levels. A decrease in extracellular glutamate levels was observed simultaneously with the onset of membrane potential recovery. Because the increase in extracellular glutamate levels during ischemia is caused by the reverse operation of glutamate transporters (Rossi et al., 2000; Mitani and Tanaka, 2003), it has been hypothesized that intraischemic hypothermia can block this reverse operation. On the contrary, decreases in extracellular glutamate levels are caused by the forward operation of glutamate transporters that use the Na+ electrochemical gradient as a driving force, which is generated by Na+/K+-ATPase (Barbour et al., 1988; Nicholls and Attwell, 1990; O'Kane et al., 1999). Accordingly, the decrease in extracellular glutamate levels is an energy-dependent process, and the recovery of membrane potential is essential for this decrease.
In the present study, the rates of decrease in extracellular glutamate levels were the same in the normothermia (−36.6 μmol·L−1·min−1) and intraischemic hypothermia (−36.0 μmol·L−1·min−1) groups. These observations suggest that although intraischemic hypothermia blocked an increase in extracellular glutamate levels, it did not disrupt their decrease. Increases and decreases in extracellular glutamate levels primarily depend on the reverse and forward operations of glutamate transporters, respectively, under ischemic conditions (Rossi et al., 2000; Mitani and Tanaka, 2003). Because it has been reported that reverse operation does not reflect the process of forward operation (i.e., differences in the degree of Na+ voltage dependency in the site of the K+ relocation branch) (Zhang et al., 2007; Grewer et al., 2008), the sensitivity of each process to hypothermia appears to be different. Thus, intraischemic hypothermia can be safely implemented without disrupting decreases in extracellular glutamate levels.
The CBF thresholds for membrane potential recovery were significantly lower in the intraischemic hypothermia group than in the normothermia group (p = 0.02). Extracellular glutamate levels at this point were also significantly lower in the intraischemic hypothermia group than in the normothermia group (p = 0.004). Because the glutamate transporter forms a macromolecule with Na+/K+-ATPase and is functionally linked to it (Rose et al., 2009), the energy necessary for membrane potential recovery is lower under low extracellular glutamate levels. In clinical situations, the early initiation of intraischemic hypothermia blocks an increase in extracellular glutamate levels and thus facilitates membrane potential recovery.
This study has several limitations. First, CBF was decreased by a rate of 2.5% of the baseline per minute by draining blood. This gradual decrease in CBF did not mimic the actual changes occurring in CBF during cardiac arrest. Although cerebral ischemia suddenly occurs after cardiac arrest, the metabolic changes in neuronal cells gradually progress for ∼2 minutes until the onset of membrane potential loss. A gradual change in CBF was necessary for accurate determination of the target temperature threshold for suppressing the increase in extracellular glutamate levels and for determination of the CBF threshold for membrane potential recovery.
Second, in the present study, we performed selective brain cooling using nasopharyngeal cooling, and the brain temperature was measured in the epidural space. Since it has been reported that the epidural temperature is lower than the subcortical temperature by 1.0°C during the nasopharyngeal cooling of rats (Hagioka et al., 2003) and primates (Takeda et al., 2012), we may need to decrease the epidural temperature by a further 1.0°C to protect subcortical regions. On the contrary, in whole body cooling, it seems that differences in temperature between cortical and subcortical regions are relatively small. However, since it has been reported that subcortical temperature is higher than the rectal or esophageal temperature by 0.34°C (Coppler et al., 2016), body temperature may also need to be controlled to compensate for this difference.
Third, in the present study, the target temperature for suppressing increases in extracellular glutamate levels was observed. However, this could be different from the target temperature for suppressing neuronal damage, since there is a clear difference in the mechanisms of neuronal damage between the ischemic period and the recirculation period (Dirnagl et al., 1999). For example, the formation of oxygen free radicals is observed only during the recirculation period and is reported to be suppressed by lowering brain temperature to less than 30°C (Globus et al., 1995). In contrast, as shown in the present study, glutamate is released only during the ischemic period. The target temperature for suppressing neuronal damage would be different between the ischemic period and the recirculation period.
Conclusions
The target temperature for blocking glutamate release during intraischemic hypothermia was found to be 32.6°C ± 0.9°C (95% confidence interval: 32.0–33.2°C). Our results suggest that earlier induction of intraischemic hypothermia can maintain low glutamate levels without disrupting glutamate reuptake. Membrane potential recovery is essential for a decrease in extracellular glutamate levels. Because earlier initiation of intraischemic hypothermia can suppress extracellular glutamate levels, the CBF threshold for membrane potential recovery can also be lowered.
Footnotes
Acknowledgments
This study was supported by a grant from the Ministry of Health, Labour and Welfare of Japan (26293345) and a grant from Daiken Medical Co. No funding sources were involved in the study design; collection, analysis, and interpretation of the data; writing of the article; or in the decision to submit the article for publication.
Author Disclosure Statement
Dr. Takeda is developing a pharyngeal cooling system to decrease the brain temperature during resuscitation, particularly before the return of spontaneous circulation. His institution received grant support and royalties from Daiken Medical. No competing financial interests exist for the remaining authors.