
Abstract
Stroke is the second leading cause of death worldwide; nevertheless, its pathological mechanism still remains unclear. Besides, there is an urgent need to find new effective treatment strategies for patients with stroke. Hypothermia therapy is widely used in clinical as a neuroprotective strategy practice. However, the exact mechanism is not fully understood. In this study, we examined the effects of hypothermia on glial glutamate transport-1 (GLT-1) and the extracellular glutamate concentration ([Glu]e) in cerebral ischemia–reperfusion insult rats. Our results revealed that cerebral brain ischemia–reperfusion caused the decrease of GLT-1 and Bcl-2, the increase of Bax and [Glu]e, and caused neuron loss. On the contrary, head mild hypothermia (HMH) for 2 hours alleviated the abovementioned effects and exerted neuroprotection. In the hypothermia group, pretreatment with dihydrokainate, a functional antagonist of GLT-1 by lateral ventricle injection partly reversed the abovementioned effect of HMH. Our results suggest that HMH could exert a neuroprotective role by maintaining GLT-1 and reducing the excitotoxicity of [Glu]e during ischemia–reperfusion insult in rats.
Introduction
Stroke is the second leading cause of death worldwide and the leading cause of mortality in China (Zhou et al., 2016; Feigin et al., 2019). Recently, some new treatment strategies have been proposed for patients with stroke, including drug therapy (Ginsberg, 2008), ischemic preconditioning (Zhang et al., 2017), exercise preconditioning (Yang et al., 2012), and hypothermia (Hagioka et al., 2003). Busto et al. (1987) have proposed the use of hypothermia for cerebral infarction patients in 1987. Since then, many basic studies (D'Ambrosio et al., 2013; Ma et al., 2016) and clinical trials (Su et al., 2016; Olah et al., 2018) have confirmed that hypothermia can effectively improve the prognosis of patients with cerebral ischemic diseases.
Glutamate (Glu) is a major excitatory amino acid in the mammalian brain, which not only acts as a neurotransmitter of excitatory signals (Willard and Koochekpour, 2013; Parkin et al., 2018), but also as a neurotoxin. Excessive extracellular Glu results in excitotoxicity and neuronal apoptosis (Karki et al., 2018). Neurotransmission abnormalities occur in many brain diseases and are associated with abnormal concentrations of Glu (Shigeri et al., 2004). Glial glutamate transport-1 (GLT-1) has a major role in maintaining a low concentration of glutamate in the extracellular space (Rothstein et al., 1996; Rao et al., 2001). However, so far, few studies have reported on the relationship between GLT-1 and the neuroprotective effect of hypothermia during cerebral ischemia–reperfusion. Thus, the aim of this study was to investigate the effect of head mild hypothermia (HMH) on GLT-1 and the extracellular glutamate concentration ([Glu]e) after global cerebral ischemia–reperfusion in rats, to further explore the neuroprotective mechanism of mild hypothermia.
Materials and Methods
Animals and grouping
Adult male Wistar rats (220–250 g) were obtained from The Experimental Animal Center of Hebei Medical University. All the animals were housed in an environment at a temperature of 22°C ± 1°C, relative humidity of 50% ± 1%, and a light/dark cycle of 12/12 hours, with free access to food and water. All animal studies (including the mice euthanasia procedure) were carried out in compliance with the regulations and guidelines of Hebei Medical University institutional animal care and conducted according to the AAALAC and the IACUC guidelines (Animal use protocol No. 1509027).
Before each experiment, all rats were acclimatized to the laboratory environment for 7 days.
Experiment 1: changes in GLT-1 in rats during the cerebral ischemia–reperfusion insult
A total of 180 rats were randomly divided into three groups (Supplementary Fig. S1): control group (C, n = 12), sham group (S, n = 84), and ischemia–reperfusion group (I/R, n = 84). The animals in sham and ischemia–reperfusion groups were killed and sampled at time points (after reperfusion for 0, 8, and 16 hours, and 1, 3, 5, and 7 days, n = 12 in each subgroup) and animals in control group were killed and sampled directly. Brains of each subgroup were used for the assay of GLT-1 protein in hippocampal CA1 region by immunohistochemistry (n = 6) and western blot analysis (n = 6).
Experiment 2: effect of HMH on GLT-1 in rats during cerebral ischemia–reperfusion injury
Previous studies have confirmed that nasopharynx cavity cooling has a neuroprotective effect (Hagioka et al., 2003; Takata et al., 2005). To observe the effect of HMH on the reduce of GLT-1 caused by ischemia–reperfusion insult, we added the following three groups on the basis of experiment 1: hypothermia ischemia–reperfusion group (HI/R group, n = 12): occlusion of bilateral vertebral arteries (OBVAs) was performed for 48 hours, after which the bilateral common carotid arteries (BCCAs) were blocked for 8 minutes after the hippocampal temperature dropping to 32°C through nasopharynx cavity cooling method, keeping hypothermia for 2 hours; normal saline (NS)+HI/R group (n = 12): 20 μL of 0.9%NS was intracerebroventricularly injected 30 minutes before ischemia operation, other procedures were the same as HI/R group; dihydrokainate (DHK)+HI/R group (n = 12): 20 μL of DHK was injected into lateral ventricle 30 minutes before ischemia operation, other procedures were the same as HI/R group. Considering that GLT-1 was obviously decreased 7 days after the operation (experiment 1), only the GLT-1 at 7 days was used for further analysis (Supplementary Fig. S1).
Experiment 3: effect of GLT-1 on the [Glu]e in rats during global cerebral ischemia–reperfusion injury
HMH can reduce the extracellular glutamate concentration during and after cardiac arrest in rats (Takata et al., 2005). To clarify the correlation between GLT-1 and hypothermia neuroprotective, we observed the changes of [Glu]e. In this experiment, 30 rats were randomly divided into 5 groups: sham group (S, n = 6), ischemia–reperfusion group (I/R, n = 6), HI/R group (n = 6), NS+HI/R group (n = 6), and DHK+HI/R group (n = 6). Treatments of animals were the same with experiments 1 and 2. The extracellular fluid from the hippocampal CA1 region was collected by microdialysis at various given periods (Fig. 5A), and the [Glu]e was analyzed by high-performance liquid chromatography (HPLC).

Measurement of the [Glu]e.
Establishment of global cerebral ischemia–reperfusion model
In this study, four-vessel occlusion (4-VO) (Zhang et al., 2017) was used to establish an animal model of global cerebral ischemia–reperfusion. First, the bilateral vertebral arteries were electrocauterized under isoflurane anesthesia. After 48 hours of recovery, the BCCAs of the rats were exposed under isoflurane anesthesia and procaine (1%) local anesthesia. After the rats recovered from the anesthesia, the BCCA was clamped for 8 minutes to induce ischemic brain insult. Animals that were not conscious, with no righting reflex and pupils dilated after the occlusion, were used as cerebral ischemia models. The wounds were sutured after each operation. In the sham group, OBVA was performed, after which the BCCAs were exposed without blocking the blood flow.
Nasopharyngeal cavity cooling method
Endotracheal intubation with ventilation was performed after isoflurane anesthesia. A temperature probe was inserted into the right hippocampus. An 18G intravenous catheter was inserted into the nasopharynx of each rat, fixed, and a block of cotton was placed in the throat to prevent aspiration. Cool 0.9%NS (4°C) was pumped into the nasopharynx at a rate of 100 mL·kg−1·min−1, and continuous suction was simultaneously maintained. The speed was adjusted according to the thermometer data to maintain the hippocampal temperature at 33°C ± 0.5°C. An electric blanket was used to keep the body warm and maintain the anal temperature at 37°C ± 0.5°C (Hagioka et al., 2003).
Neuropathological evaluation
At the determined time points, the rats were deeply anesthetized and perfused through the ascending aorta with NS, followed by 4% paraformaldehyde. Coronal brain slices were stained with thionin for neuropathological evaluation. According to the methods described by Kitagawa et al. (1990) and Kato et al. (1991), the neuronal density (ND) was determined by counting the number of surviving pyramidal neurons with an intact cell membrane, a full nucleus, and a clear nucleolus within 1 mm of the CA1 region. The average number of the pyramidal neurons in six such areas in three sections from each side was calculated as the final ND value.
Collection of extracellular fluid and measurement of the [Glu]e
Rats were placed in a stereotaxic apparatus after anesthesia. A microdialysis probe (MAB 6.14.2.ss, MAB Sweden) was implanted into the parietal cortex for 2 mm (3 mm posterior to and 3 mm to the left of the bregma) and perfused with Ringer's solution at a flow rate of 2 μL/min using an infusion pump. When the electroencephalogram was stabilized, microdialysis fluid was collected every 10 minutes for 60 minutes. t0: 20–10 minutes before global cerebral ischemia, t1: 10–0 minutes before ischemia; t2: 0–10 minutes after ischemia; t3: 10–20 minutes after ischemia; t4: 20–30 minutes after ischemia; and t5: 30–40 minutes after ischemia (Fig. 5A). The collected samples were protected from light and stored at −80°C for testing. The peak areas of Glu standard products with various concentrations were determined by HPLC analyzer (Thermo UltiMate 3000, Thermo), and the standard curve was drawn according to the functional relationship between the absorption peak area and the content. According to the regression equation of the standard curve, the actual [Glu]e in rat brain tissue was calculated. The average concentration at t1 and t2 was taken as the basic value. The ratio of [Glu]e to the basic value [Glu]r was calculated for intergroup comparison (Zhang et al., 2005).
The complete experimental procedures of immunohistochemistry and western blot are given in the Supplementary Data.
Statistical analysis
Statistical analysis was performed using SPSS 21.0. All data are presented as the mean ± standard error of the mean. Comparisons were performed by one-way analysis of variance and the least significant difference method for multiple comparisons. Unmatched dates were analyzed by the nonparametric rank-sum test combined with the Dunn–Bonferroni test for multiple comparisons method to test the differences between the groups. p < 0.05 was considered statistically significant.
Results
Ischemia–reperfusion insult reduced GLT-1 in the hippocampal CA1 region, which was inhibited by HMH
First, we compared the trend of GLT-1 in the hippocampal CA1 region of the group C and group S by western blot (Fig. 1A). Compared with group C, GLT-1 in group S was significantly increased at all time points (p < 0.05); the highest level was 8 hours and 3 days after the operation. Compared with group S, GLT-1 in group I/R peaked at 0 hour, then gradually decreased (p < 0.05). Because GLT-1 was most significantly reduced on day 7, we compared GLT-1 in each group at 7 days in experiment 2 (Fig. 1B). Compared with group I/R, GLT-1 decrease in group HI/R was partly reversed at 7 days (p < 0.05), whereas no significant difference was observed between DHK+HI/R group or NS+HI/R and HI/R group (p > 0.05).
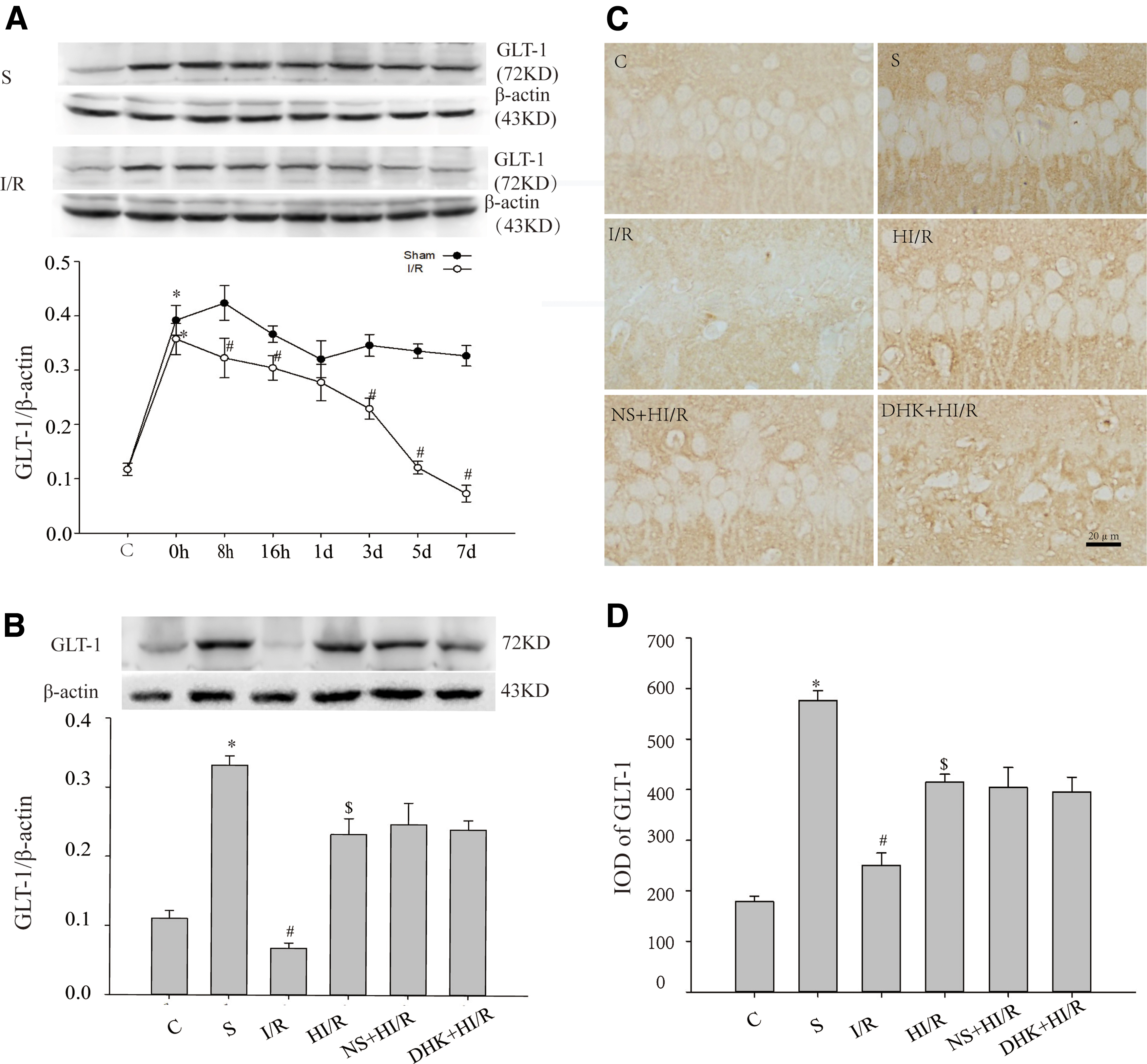
GLT-1 protein in hippocampus CA1 of rats in each group.
The above result was further confirmed using immunohistochemistry (Fig. 1C, D). GLT-1 is mainly expressed in astrocytes in the hippocampal CA1 region (Zhang et al., 2007). A small amount of GLT-1 was uniformly distributed throughout the hippocampal CA1 region in group C. In group S, the GLT-1 protein reached the highest level at 8 hours, large amounts of brown-stained GLT-1-immunopositive particles were observed in the hippocampal CA1 region, after which the leveal gradually decreased. The GLT-1 in group S was significantly higher at each time point compared with group C (p < 0.05). In I/R group, GLT-1 peaked at 0 hour, then decreased, and there was a large area of the hippocampal CA1 region in which GLT-1 expression was deficient at 7 days; GLT-1 level at each time point, except for 0 hour, was lower compared with group S (p < 0.05). Compared with the I/R group, GLT-1 increased in the HI/R group at 7 days (p < 0.05). Compared with group HI/R, GLT-1 in group DHK+HI/R and group NS+HI/R was not significantly different at 7 days (p > 0.05).
These results suggest that ischemia–reperfusion insult reduces GLT-1 in the hippocampal CA1 region, which can be inhibited by HMH.
HMH inhibited Bcl-2 decrease and Bax increase caused by ischemia–reperfusion insult, whereas DHK partially reversed this effect
Bcl-2 was mainly distributed in the cytoplasm and neuronal synapses in the hippocampal CA1 region (Fig. 2A). Some Bcl-2 was observed in group C, and there was a significant difference between group C and group S at 7 days (p > 0.05). Compared with group S, Bcl-2 in I/R group decreased (p < 0.05); compared with group I/R, Bcl-2 in the HI/R group increased (p < 0.05). Compared with the HI/R group, Bcl-2 in the DHK+HI/R group decreased (p < 0.05), whereas there was no significant difference in the NS+HI/R group (Fig. 2A, B, p > 0.05). The western blotting analysis was consistent with the result obtained by immunohistochemistry (Fig. 2C).
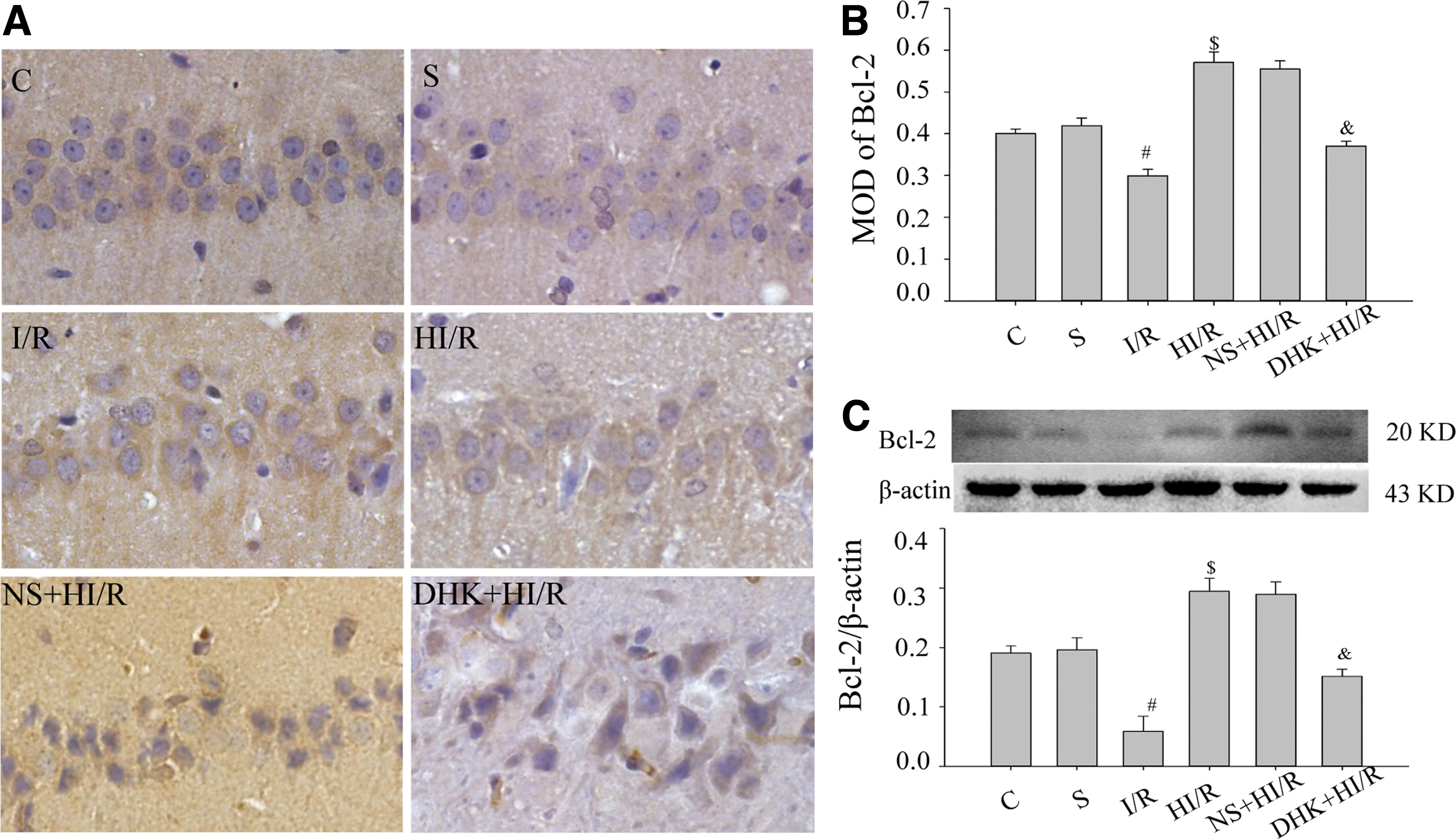
Bcl-2 protein in the hippocampal CA1 region at 7 days in each group.
Bax was also distributed in the cytoplasm and neuronal synapses. Some yellowish immunoreactivity was observed in group C (Fig. 3A). There was no difference in Bax protein between the S group and the C group at 7 days (p > 0.05). Compared with the S group, Bax in I/R group was increased (p < 0.05); compared with the I/R group, Bax in the HI/R group was decreased (p < 0.05). There was no difference between NS+HI/R group and HI/R group (p > 0.05), whereas Bax in the DHK+HI/R group was increased compared with that in the HI/R group (Fig. 3A, B, p < 0.05). The western blotting analysis was consistent with the results obtained by immunohistochemistry (Fig. 3C).
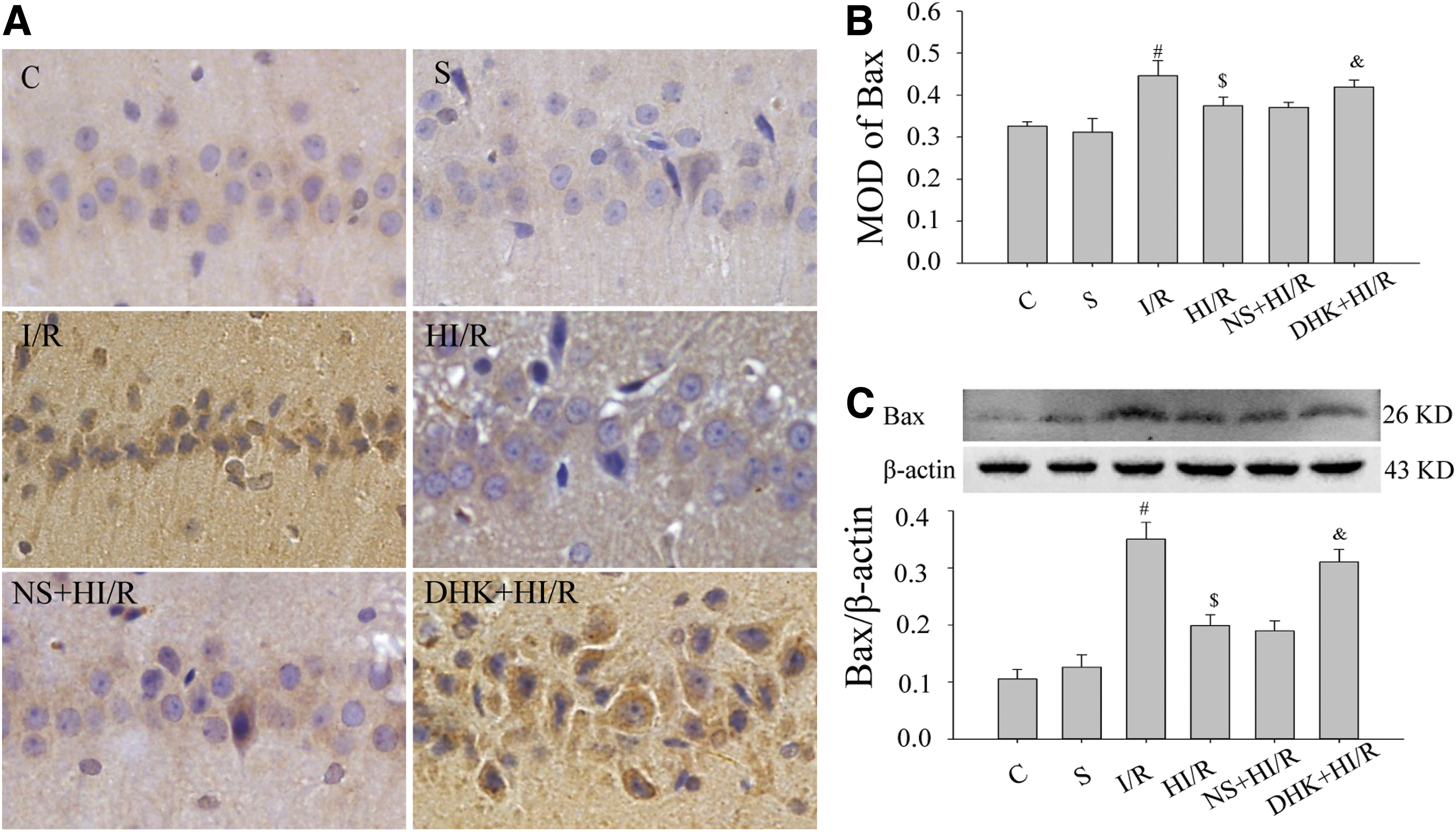
Bax protein in the hippocampal CA1 region at 7 days in each group.
These results suggest that HMH inhibits Bcl-2 decrease and Bax increase caused by ischemia–reperfusion insult, whereas DHK can partially reverse this effect.
HMH reduced neuronal loss in the hippocampal CA1 region caused by ischemia–reperfusion injury, which was partly reversed by DHK
To observe the role of GLT-1 in neuroprotection of hypothermia, we observed neuron damage in hippocampus CA1 at 7 days by thionin staining (Fig. 4). In the hippocampal CAl subfield of the C group, pyramidal neurons were arranged in order; there were 2–3 cell layers, the membranes of the neurons were intact, the nucleus was full, and the nucleolus was clear (ND: 214 ± 9.0). At 7 days, no obvious neuronal death was observed in the S group (ND: 195 ± 14.5), and no significant difference was observed compared with the C group (p > 0.05). In group I/R, there was no obvious neuronal death at 1 day; partial neuronal death was observed at 3 days (date not shown), and a large amount of neuronal death at 7 days (ND: 41 ± 8.5). The ND increased in the HI/R group (ND: 141 ± 16.0, p < 0.05) compared with the I/R group at 7 days. The ND decreased in the DHK+HI/R group compared with the HI/R group at 7 days (p < 0.05), whereas there was no significant difference in the ND between NS+HI/R group and HI/R group (p > 0.05). These results suggest that HMH inhibits neuronal loss in the hippocampal CA1 region caused by ischemia–reperfusion insult exerting a neuroprotective effect, which can be partially reversed by DHK.

Neuropathological evaluation after thionin staining of the hippocampal CA1 region in each group at 7 days.
HMH inhibited the increase in the [Glu]e caused by ischemia–reperfusion injury, which was partly reversed by DHK
To further explore the function of GLT-1 in the neuroprotective effect of HMH, [Glu]e was detected by HPLC (Fig. 5). Compared with the baseline values, there was no significant difference in [Glu]e at each given period in the S group (p > 0.05). [Glu]e in other groups at t2 and t3 were higher than baseline values (p < 0.05). [Glu]e at t4 and t5 in each group were not significantly different from baseline values (p > 0.05).
To eliminate the differences of [Glu]e in the preoperation in each rat, the ratio of the [Glu]e to the baseline ([Glu]r) was used for comparison among groups. At t0 and t1, there was no significant difference in the [Glu]r among groups (p > 0.05). Compared with the S group, [Glu]r in the I/R group increased at t2 and t3 (p < 0.05); compared with the I/R group, [Glu]r in the HI/R group decreased (p < 0.05). Compared with group HI/R, [Glu]r in the DHK+HI/R group increased (p < 0.05), whereas there was no significant difference in the NS+HI/R group (p > 0.05). Moreover, there was no significant difference at t4 and t5 among the groups (p > 0.05).
Discussion
Astrocytes, the main glial cells in the brain, have an important role in neuronal activity and the control of homeostasis in the central nervous system (CNS) (Björklund et al., 2008; Boison et al., 2010). Because astrocytes are close to capillaries, the change in astrocytes is one of the earliest events after ischemia (Panickar and Norenberg, 2005). Astrocytes activate several mechanisms that may reduce neuronal damage; for example, they produce neurotrophic factors, regulate transmitter and ion concentrations, and remove excess glutamate from the extracellular environment (Aschner et al., 2002).
Glutamate (Glu) is the main excitatory transmitter in CNS (Willard and Koochekpour, 2013; Parkin et al., 2018). High concentrations of extracellular Glu ([Glu]e) lead to the excessive activation of glutamate receptors and excitotoxicity in nerve cells (Karki et al., 2018). Many neurological diseases are associated with Glu excitotoxicity (Lewerenz and Maher, 2015; Amorini et al., 2017; Bi et al., 2017).
In this study, we observed that cerebral ischemia insult increases [Glu]e and neuronal death, which can be partially inhibited by HMH. High concentrations of [Glu]e can cause opening of voltage-gated Ca2+ channels and Ca2+ influx through activation of iGluRs causing nerve injury (Uchino et al., 2001; Lang et al., 2011; Ahmed et al., 2017), which triggers a series of downstream reactions, including ATP synthesis dysfunction, caspase, calcium-dependent proteases, and nucleotide endonuclease activation, ultimately leading to neuronal injury and death (Szydlowska and Tymianski, 2010). Some researchers have demonstrated that hypothermia significantly reduces the release of excitatory toxins and Ca2+ influx caused by cerebral ischemia (Matsumoto et al., 1993). Protecting neurons through the GluR2 receptor delayed calcium influx is one of the underlying neuroprotective mechanisms of hypothermia (Colbourne et al., 2003).
Two reasons that may explain the increase in extracellular glutamate. First, in the early stage of cerebral ischemia and hypoxia, because of energy metabolism disorder, extracellular adenosine increases (Li et al., 2001; Fredholm, 2007; Martin et al., 2007), and then promotes glutamate release from glial cells through A2a adenosine receptor (Li et al., 2001). Besides, high extracellular K+ mediated by ischemia and hypoxia can cause reverse glutamate transport (Levy et al., 1998). The second is the reduction of glutamate reuptake by cells. The brain lacks glutamate metabolic enzyme, and the low [Glu]e is mainly maintained by the glutamate transport system, also known as excitatory amino acid transporters, which can uptake Glu into glial cells and neurons. GLT-1 provides the majority of glutamate transport essential for maintaining low [Glu]e (Rothstein et al., 1996; Rao et al., 2001).
In this study, sham operation upregulated GLT-1. The occlusion of the vertebral artery in the sham group was equivalent to a mild ischemic insult. Mild ischemic preconditioning (3 minutes) can upregulate GLT-1 expression by activating the p38/mitogen-activated protein kinase (MAPK) pathway, thus increasing Glu reuptake and enhancing ischemic tolerance (Zhang et al., 2017). I/R insult reduced GLT-1 and increased neuronal death, which were inhibited by hypothermia. GLT-1 levels are attributed to both the number of cells that can synthesize GLT-1 and the GLT-1 synthesis rate in each cell. The effect of hypothermia on GLT-1 levels during ischemia–reperfusion injury may be because hypothermia protects astrocytes and retains more nerve cells with GLT-1 synthetic function, thus maintaining more GLT-1. Under ischemia–reperfusion insult, neurons are more likely to be damaged, and lethal insult also causes glial cell damage (Liu et al., 2020). Studies have demonstrated that hypothermia has protective effects on astrocytes (Cao et al., 2010; Ding et al., 2014). Hypothermia may also promote the synthesis of GLT-1 in nerve cells through several intracellular signals, including transcription and translation. Researchers have found that the decrease in GLT-1 protein induced by cerebral ischemia injury is dependent on overactivation of the P38 MAPK pathway (Zhang et al., 2019), whereas hypothermia can inhibit activation of P38 (Wang et al., 2014; Yang et al., 2019).
In addition, activation of the PI3K/Akt pathway can upregulate GLT-1 expression (Zhang, 2013; Feng et al., 2014; Peng et al., 2019). Our previous study also demonstrated that hypothermia exerts neuroprotective effects through PI3K/Akt signaling, so we speculated that hypothermia may regulate GLT-1 expression through the PI3K/Akt and/or MAPK pathways. Therefore, hypothermia may maintain GLT-1 levels by preserving glial cells and/or promoting the synthesis of GLT-1 in cells after ischemia. Our research team is planning to perform a study to investigate the mechanism underlying the effect of hypothermia on GLT-1 protein levels. Given that high concentrations [Glu]e lead to the excessive activation of glutamate receptors and excitotoxicity in nerve cells (Karki et al., 2018), we speculated that hypothermia might have a neuroprotective role by maintaining GLT-1 and increasing up take of [Glu]e. To testify this effect, we used DHK, functional antagonist of the GLT-1, to inhibit the transport function of GLT-1. We found that [Glu]e and neuronal death increased, which confirmed our hypothesis.
Bcl-2 and Bax belong to the Bcl-2 gene family and are closely related to apoptosis. Bcl-2 can inhibit cell apoptosis and improve cell viability, whereas Bax promotes cell apoptosis (Shimizu et al., 2001; Gustavsson et al., 2007). We chose Bcl-2 and Bax as bioindicators to evaluate apoptosis. We found that Bcl-2 decreased, and Bax increased after I/R insult, which can be inhibited by HMH, whereas DHK treatment partially reversed the effect of HMH. The Bcl-2 gene family has a crucial role in regulating the opening and closing of permeability transition pore (PTP). PTP is a complex composed of adenine nucleotide translocator (ANT) located in the inner membrane and voltage-dependent anion channel (VDAC) located in the outer membrane and is essential for information exchange between the inner and outer mitochondria. External apoptotic stimuli, such as high concentrations of Ca2+, cause PTP opening, which may lead to mitochondrial swelling and fragmentation (He and Lemasters, 2002; Kim et al., 2012). Bax can mediate the opening of PTP through the binding of ANT or VDAC to promote cell apoptosis, whereas Bcl-2 can exert anti-apoptotic effects by competitively binding to ANT with Bax or directly preventing the binding of Bax to ANT and VDAC (Harris and Thompson, 2000). Changes of Bcl-2 and Bax expression in this study suggest that hypothermia could protect hippocampal neurons from apoptosis caused by ischemia–reperfusion insult.
This study has a few limitations. First, thionin staining cannot accurately distinguish necrosis from apoptosis. Although Bcl-2 and Bax can reflect the level of apoptosis, TUNEL and flow cytometry are more specific. Second, we only studied the protective effect of HMH on the ischemia–reperfusion injury at the cellular level and molecular level. Still, we did not evaluate the recovery of neurological function with behavioral evaluation. Finally, we observed the Glu concentration for 60 minutes during ischemia–reperfusion, which reflected early changes that result from ischemia–reperfusion but not the long-term changes.
Conclusion
Cerebral ischemia–reperfusion decreases GLT-1 and increases [Glu]e, which produces excitatory toxicity and leads to neuron death in the hippocampal CA1 region. Mild head hypothermia can promote extracellular Glu reuptake by maintaining GLT-1. Furthermore, it can reduce excitotoxicity, inhibit apoptosis, and exert a neuroprotective role. Consequently, GLT-1 might be a potential target for the treatment of cerebral ischemia–reperfusion injury and can better guide the clinical application of mild hypothermia.
Footnotes
Acknowledgments
This study was performed at Department of Pathophysiology, Hebei Medical University and Hebei Key Laboratory of Critical Disease Mechanism and intervention. The authors thank Professor Wenbin Li, director of Pathophysiology Department of Hebei Medical University, for his valuable technical assistance and provision of research platform. This article has been preprinted in Research Square (doi: 10.21203/rs.2.17383/v1).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by Natural Science Foundation of Hebei Province, China (No: H2018206345).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.