
Abstract
Background:
Iodide transport defects (ITDs), rare causes of congenital hypothyroidism (CH), have been shown to arise from abnormalities of the sodium/iodide symporter (NIS). We describe a 16-year-old girl with CH caused by an ITD resulting from a novel mutation of NIS.
Summary:
A 16-year-old girl with CH diagnosed by a neonatal screening program received early treatment with L-thyroxine replacement therapy. A 123I scan had failed to reveal any iodide uptake by the thyroid and salivary glands; thus, thyroid agenesis was diagnosed. Thyroglobulin (Tg) was not measured when she was a neonate or infant. Unexpectedly, at the age of 14.5 years, a nodular goiter and high serum Tg concentrations (303 ng/mL; normal, <50) were identified. Her thyroid radioactive iodine uptake was very low as was the saliva to plasma iodide ratio (0.5). Analysis of her NIS gene revealed an in-frame six-nucleotide deletion of the coding sequence (1206-1211delGTCGGC) corresponding to the deletion of amino acids 287 and 288 of the human NIS protein located at the beginning of the VIII transmembrane segment. The proband was homozygous for this deletion, whereas both unrelated parents and her brother were heterozygous. COS-7 cells transfected with the mutant NIS failed to concentrate iodide, confirming that the mutation was the direct cause of the ITD in this patient.
Conclusions:
We describe a patient with CH caused by a previously not described mutation of the NIS gene that was inherited from her parents. We therefore recommend that thyroid ultrasonography be performed in CH patients with low radioactive iodine uptake and elevated serum Tg.
Introduction
The human NIS (hNIS) gene is localized on chromosome 19p12-13 and consists of 15 exons, encoding a glycoprotein of 643 amino acids (9). The NIS is predicted to have a serpentine structure with 13 transmembrane domains with an amino-terminus facing the extracellular milieu and an intracellular carboxyl terminus facing the cytosol (7). To date, several loss-of-function mutations of the NIS gene and two deletions (19,20) have been described in patients with ITD (10 –18). In this article, we describe a new homozygous deletion of two amino acids in the NIS protein in a patient with CH having the complete clinical picture of ITD.
Patients and Methods
Case report and family members
The proband was a girl who was detected to have CH by a neonatal screening program. She was born at term by caesarean section after an uncomplicated pregnancy to unrelated healthy Italian parents. Her birth weight was 2550 g and her length 51 cm. Her neonatal history recorded 15 days of jaundice. The diagnosis of CH was confirmed by serum thyroid function tests: thyrotropin (TSH) 506 mU/L (normal range, 0.5–4.2) and free thyroxine (FT4) 1.2 pg/mL (normal range, 7–17). L-thyroxine (L-T4) replacement therapy was started immediately after the first evaluation at 20 days of age at an initial dose of 8 μg/kg · day. Tg was not measured as a neonate or infant. Replacement therapy was modified during follow-up according to clinical and hormonal evaluation to maintain normal serum TSH and serum FT4 in the upper normal range. At 3 years of age, after temporary withdrawal of L-T4 therapy for 6 weeks, a 123I thyroid scan failed to show any thyroid tissue in the neck or base of the tongue. It was concluded that the child had thyroid gland agenesis. The diagnosis of primary hypothyroidism was confirmed by the finding of high serum TSH and low FT4 concentrations (TSH 180 mU/L and FT4 0.13 pg/mL); thus, L-T4 replacement therapy was soon re-started.
Physical and mental development proceeded normally. Menstrual cycles were regular since menarche age 14, and her final height was appropriate to her genetic target (155 cm). She completed her high school education.
At the age of 14.5 she developed a neck mass. A nodular goiter was present at thyroid ultrasonography showing an enlarged, normally located gland with two dominant nodules. A high serum TSH (14.8 mU/L) and Tg (303 ng/mL; normal, <50) concentration with a normal FT4 concentration (10 pg/mL) was present. Poor compliance with L-T4 treatment in the years before the last evaluation was suspected and later confirmed by the patient. The clinical finding of CH with goiter together with high plasma Tg level and absent thyroid scan uptake suggested the diagnosis of an iodide-trapping defect. The patient was admitted to the Department of Endocrinology at the University of Pisa when she was 16 years old. The proband's parents and her older brother, who were clinically euthyroid, were studied. The patient, the parents, and the brother gave informed consent to undergo studies approved by the local ethics committee.
Laboratory and thyroid ultrasonography evaluation
Serum FT4 and free triiodothyronine (FT3) concentrations were determined by a chemiluminescent method (Vitros System, Ortho-Clinical Diagnostics, Johnson & Johnson, and Amersham, Bucks, United Kingdom; normal ranges, 7–17 and 2.7–5.7 pg/mL, respectively). Serum TSH was measured using an ultrasensitive commercial chemiluminescent method (Immulite 2000; Diagnostic Products, Los Angeles, CA; normal range, 0.4–3.4 μU/mL). TPO and Tg antibodies (Abs) were measured using a two-step immunoenzymatic assay (AIA-Pack TgAb, and TPOAb; Tosoh, Tokyo, Japan) and expressed as U/mL. Normal values were <30 U/mL for TgAb and <10 U/mL for TPOAb. Serum Tg was measured using an immunometric chemiluminescence assay (Immulite 2000; Diagnostic Products). Urinary iodine excretion was measured by a colorimetric assay using an autoanalyzer (Technicon, Tarrytown, NY), and results were expressed as micrograms of iodine per liter urine. Thyroid ultrasonography (US) examination was performed on the proband and on her three family members using a 7.5-MHz linear transducer (Technos, Esaote, Biomedica, Genova, Italy).
Dynamic tests performed in vivo
Thyroid radioactive iodide uptake (RAIU), measured as percentage of administered oral 131I dose (50 μCi, 1.85 MBq), was performed at 1, 2, 3, 4, 5, and 24 hours after oral administration of 131I (Atomlab 950 [PC] Biodex Medical System, New York, NY). Thyroid scan was performed with 99mTc and 99mTc-SESTAMIBI (Cardiolite; Bristol-Myers Squibb, Bruxelles, Belgium) using dual-head gamma-camera (Axis; Picker International, Highland Hts, OH) and with 131I using a dedicated one-head gamma-camera (Adac Thyrus, Aalborg, Denmark). The saliva-to-plasma (S/P) iodide ratio was measured using a modification of the method described by Harden et al. (21). Saliva was collected without stimulation over a period of 5–10 minutes, 1 hour after the oral administration of 50 μCi of 131I. At the same time, a venous blood sample was obtained and radioactivity measured in equal volumes of these fluids.
Gene sequencing
Genomic DNA was extracted from peripheral lymphocytes using standard procedures. Each exon of the hNIS gene was amplified by polymerase chain reaction using pairs of primers annealing at flanking introns. Exons 2 and 3, 6 and 7, 9 and 10, and 11 and 12 were coamplified with the intervening introns. All exons, as well as at least 15 nucleotides at all exon–intron boundaries, were sequenced in both orientations using AmpliTaqDNA polymerase FS, with an ABI Prism Bigdye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) and were analyzed on a sequencer (model ABI PRISM 310 genetic Analyser; Applied Biosystems). The oligonucleotide primers were as previously described (22).
Construction of the expression vector and functional analysis
Preparation of wild-type (wt) hNIS gene in the expression vector pcDNA3 has been previously described (12). Mutant harboring the 1206–1211del of six nucleotides was generated by site-directed mutagenesis using the Gene Tailor site-directed mutagenesis system (Invitrogen Life Technologies, Carlsbad, CA). The accuracy of the recombinant construct was verified by direct sequencing.
COS-7 cells were transfected with 500 ng of the wt hNIS or mutant hNIS, or with the empty vector by the diethylaminoethyl (DEAE)-dextran method (23). To mimic hNIS expression in the parents and the brother heterozygous for the mutation, equal amounts of wt and mutant hNIS vectors were transfected into COS-7 cells. Cotransfection using wt NIS together with the expression vector alone was performed as control.
About 200,000 COS-7 cells were seeded in 3.5 cm tissue culture plates and cultured in Dulbecco's modified Eagle's medium containing 10% fetal calf serum, 50 U/mL penicillin, 50 Fungizone, and 1 mM sodium pyruvate (Invitrogen Life Technologies). Forty-eight hours after transfection, cells were assayed for iodide uptake as described (24). Briefly, Na125I uptake was determined by incubating cells at 37°C with 1 mL buffer A (Hanks' balanced salt solution containing 0.5% bovine serum albumin and 10 mM HEPES, pH 7.4), 10 μM NaI containing about 200,000 counts/min of carrier-free Na125I in the presence or in the absence of NaClO4 1 mM. After 5-minute incubation, cells were quickly washed twice with 4 mL of ice-cold buffer A. Cells were then solubilized with 1 mL of 0.1 M NaOH, and the radioactivity was measured using a gamma-counter. Iodide uptake data were expressed as pmol/well.
Results
Current clinical features and investigation
At the age of 16 the proband underwent clinical and morphological examinations at the Department of Endocrinology at the University of Pisa. She was 155 cm tall and weighed 52 kg. Her heart was normal by two-dimensional echocardiography, and her bone age corresponded to her chronological age. The result of ultrasonography of the abdomen was normal. On X-ray no hip abnormalities were observed.
Thyroid US confirmed the presence of a slightly enlarged thyroid gland (volume 22 mL; normal range, 5–13), with a normoechogenic pattern. Solid hypoechogenic nodules measuring 13 × 21 × 26 mm in the right lobe and 7 × 11 × 13 mm in the left lobe were found. Four other small nonpalpable hypoechogenic nodules were also noted. Fine-needle aspiration of the nodules was read as benign.
One month after L-T4 withdrawal, the serum FT4 was <1 pg/mL, FT3 was <1 pg/mL (normal range, 2.7–5.7), the TSH was >75 mU/L (normal range, 0.4–3.4), and the Tg was 657 ng/mL (normal range, <50). Serum anti-TgAb and anti-TPOAb were undetectable. Urinary iodide was 50 μg/L (normal, <300).
Dynamic tests performed in vivo
The RAIU was very low: 1.5%, 0.4%, 1.1%, 0.9%, 0.7%, and 0.3% at 1, 2, 3, 4, 5, and 24 hours, respectively (Fig. 1). Scans, performed with 99mTc (Fig. 2A) and 131I (Fig. 2B), showed no uptake in thyroid tissue in the salivary glands and stomach. A scan using 99mTc-SESTAMIBI showed normal thyroid uptake (Fig. 2C). Further, the S/P ratio was 0.5, 1, 0.9, 0.8, 0.9 and 1.1 at 1, 2, 3, 4, 5, and 24 hours, respectively (normal range, 25–140), showing that the inability of the thyroid gland to concentrate iodide was also present in the salivary glands (Fig. 3).
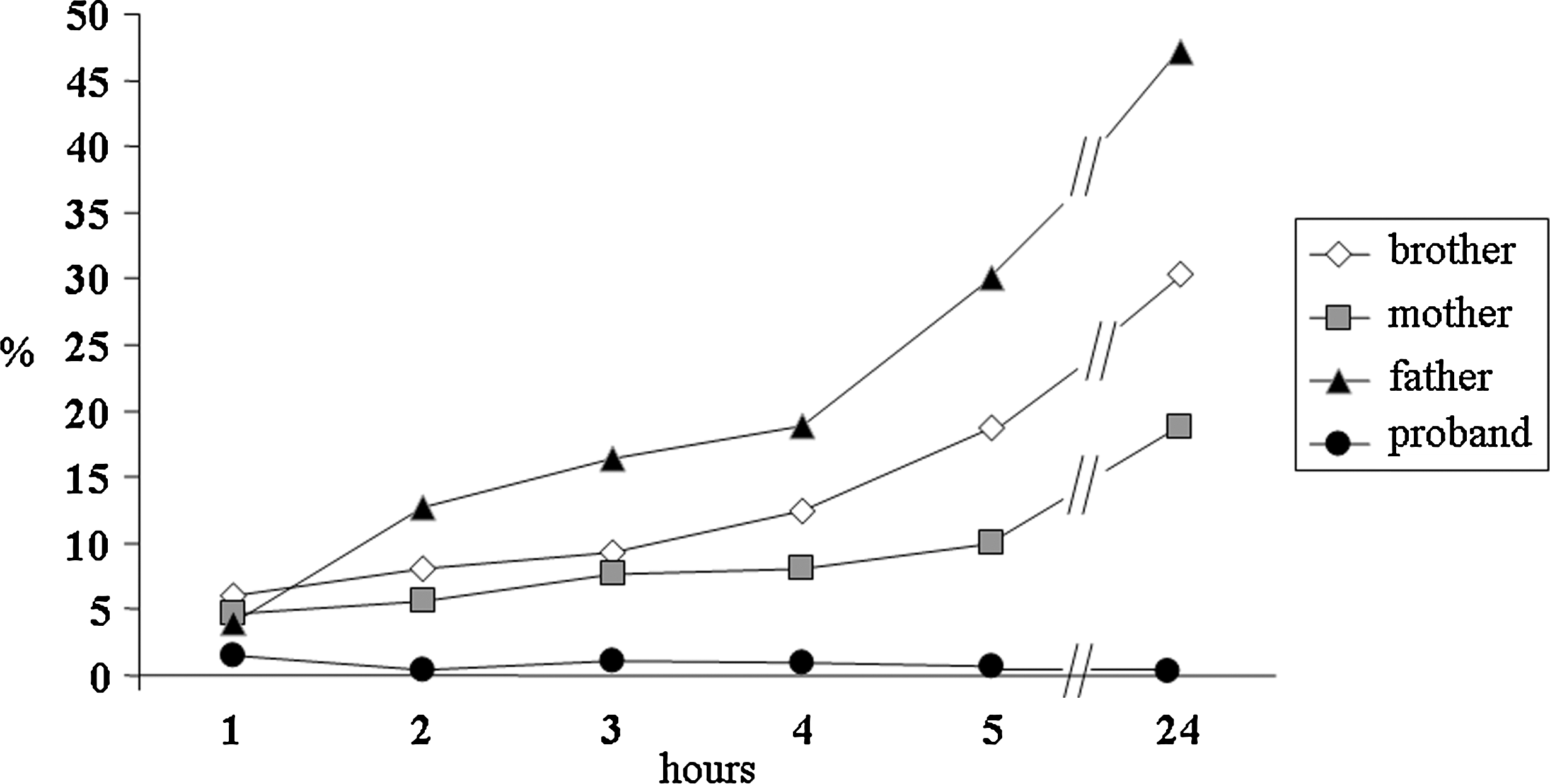
Thyroid uptake measured as percentage of administered oral 131I dose (50 μCi, 1.85 MBq) in the proband and her family members: radioiodine uptake was very low in the proband and normal in the parents and in the brother.
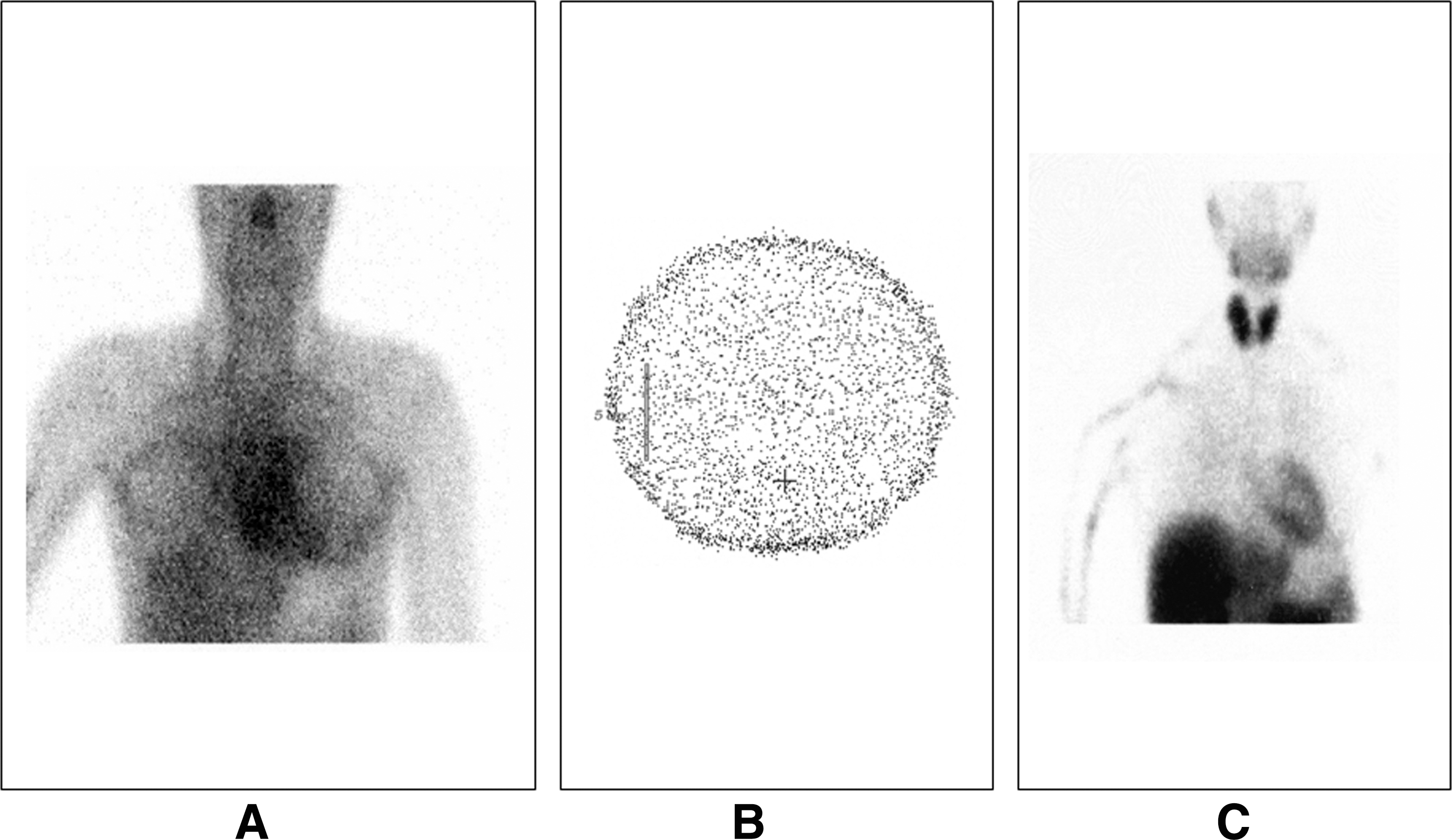
Scans performed with 99mTc and 131I failed to show thyroid tissue (
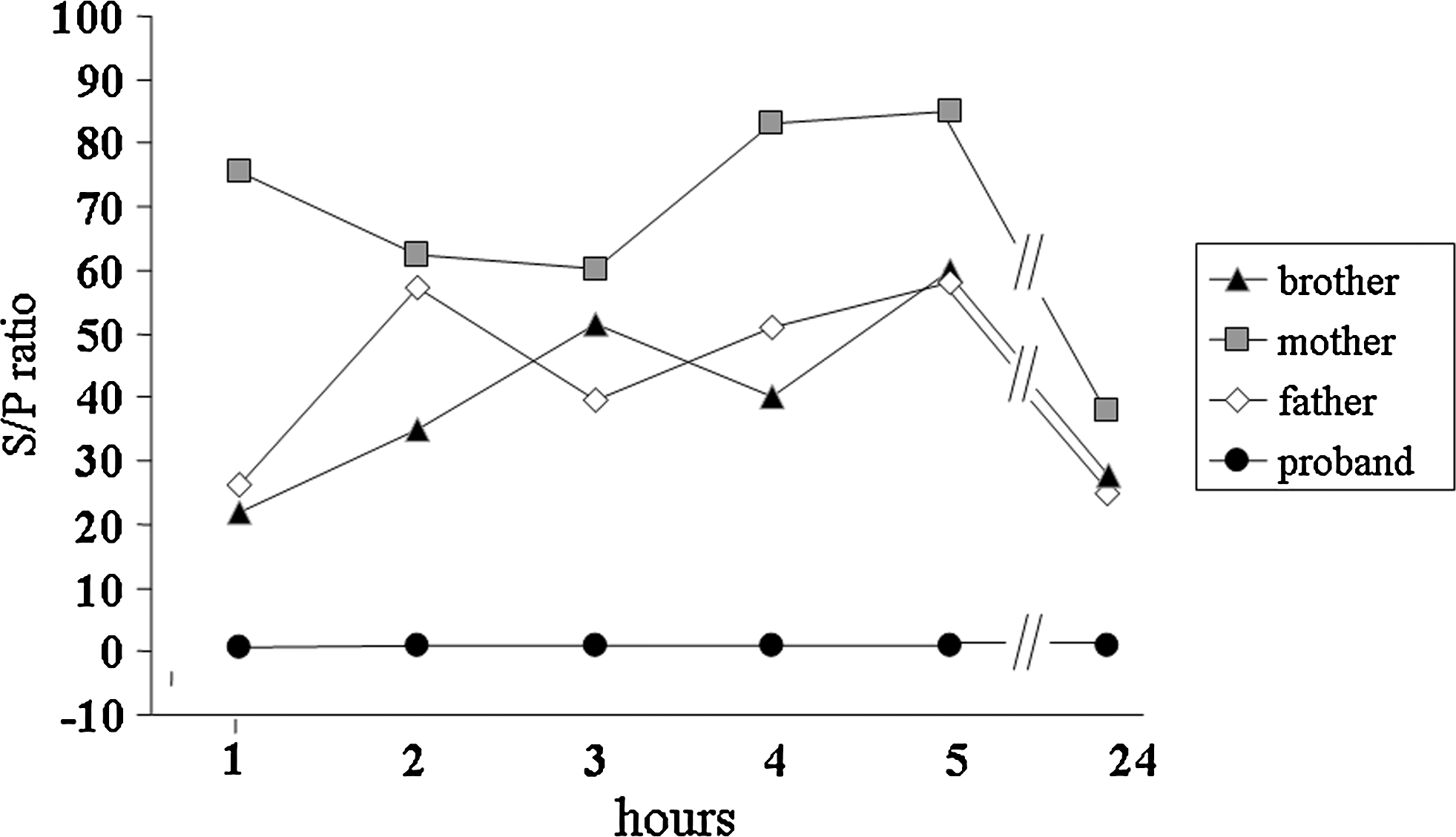
The saliva-to-plasma (S/P) iodide ratio was measured at 1, 2, 3, 4, 5, and 24 hours in the proband and in her family members. S/P ratio was always <1 in the proband, showing the inability of the thyroid gland and salivary glands to concentrate iodide, and was normal (S/P ratio >25) in the family members.
Family members
The results of the thyroid function tests (TSH, FT4, and FT3) conducted on the proband's parents and older brother were in normal range, and TPOAb and TgAb were not detectable. Her father had a small nontoxic diffuse goiter on thyroid US, and the mother and brother had normal thyroid US. The parents and older brother's RAIU was normal as was their S/P ratio (>25) (Figs. 1 and 3).
Sequencing analysis
Direct sequencing of genomic DNA of the proband revealed the presence of a six-nucleotide in-frame deletion, 1206-1211delGTCGGC, in exon 7 of the NIS gene (Fig. 4A II-2, C). The nucleotide deletion corresponded exactly to the deletion of amino acids 287 and 288 of the hNIS protein located at the beginning of the VIII transmembrane segment (Fig. 5). The patient was homozygous for this deletion (Fig. 4C), whereas both parents (Fig. 4A I-1 and I-2) and the brother (Fig. 4A II-1) were heterozygous for the same deletion (Fig. 4D).
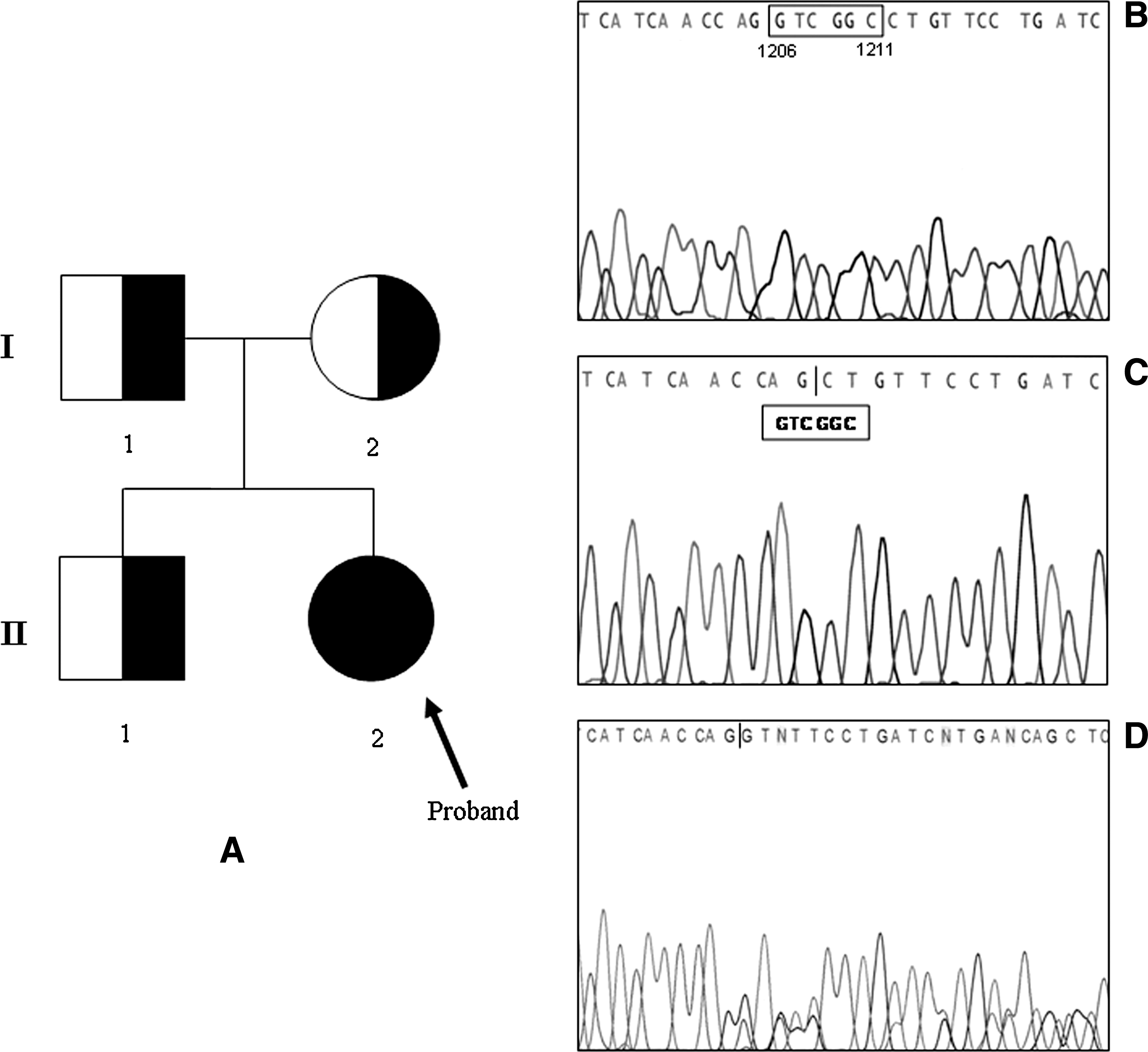
Pedigree diagram of the proband's family. Circles, females; boxes, males; arrow, proband; right filled quadrat or circlet, heterozygous deletion; full circle, homozygous deletion. Family members are indicated by generation (Roman number) and individual (Arabic number) (
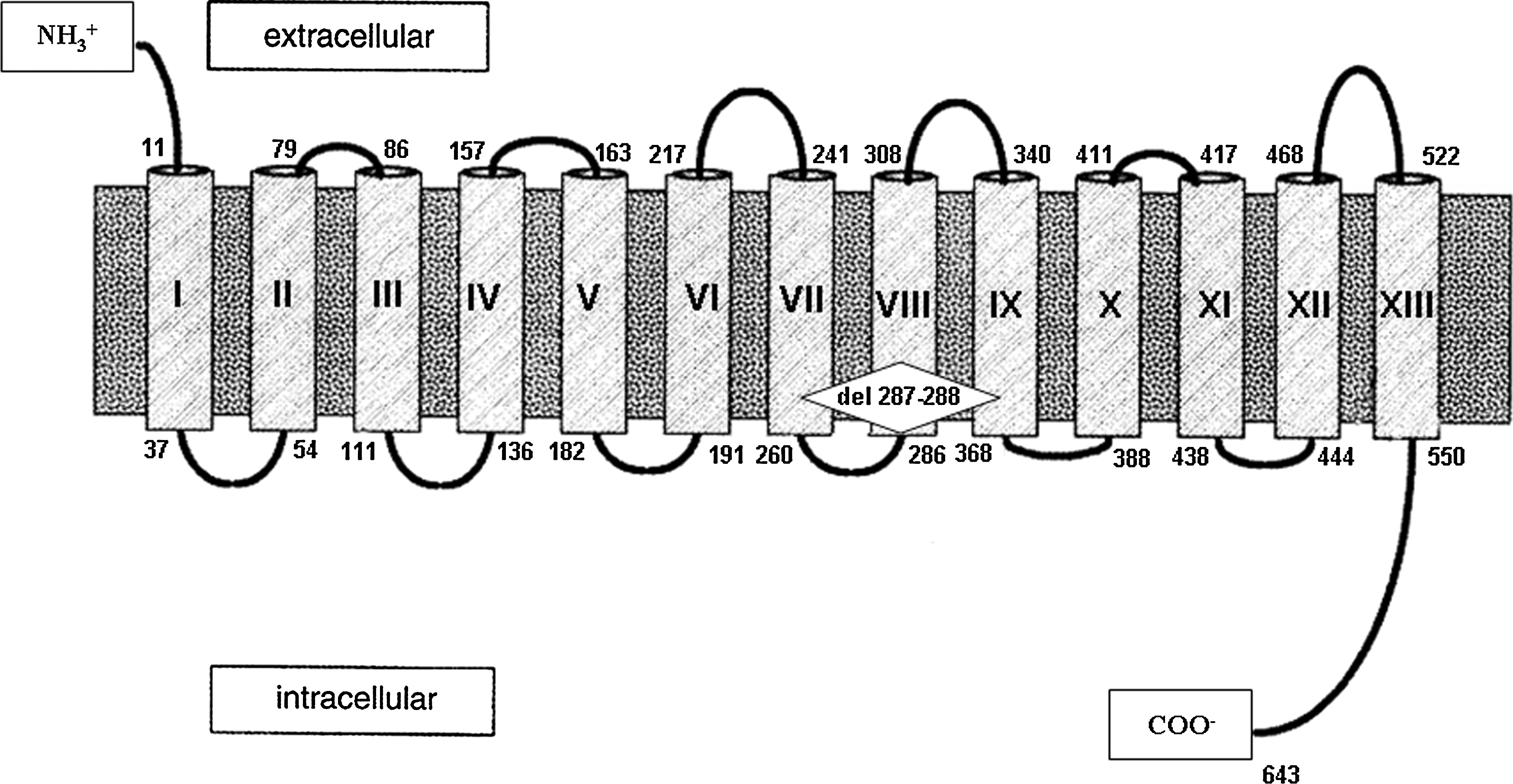
Schematic representation of the human sodium/iodide symporter (hNIS) protein and localization of NIS 1206-1211delGTCGGC.
Functional studies
COS-7 cells that were transfected with the mutant NIS 1206-1211delGTCGGC concentrated 125I similarly to cells transfected with the empty vector, suggesting that the mutation was the direct cause of the phenotype in this patient (Fig. 6). Cells cotransfected with both wt NIS and 1206-1211delGTCGGC of NIS showed a lower iodide uptake activity as compared to cells transfected with wt NIS alone, but uptake was similar to that in cells cotransfected with wt NIS and the empty vector (Fig. 6).
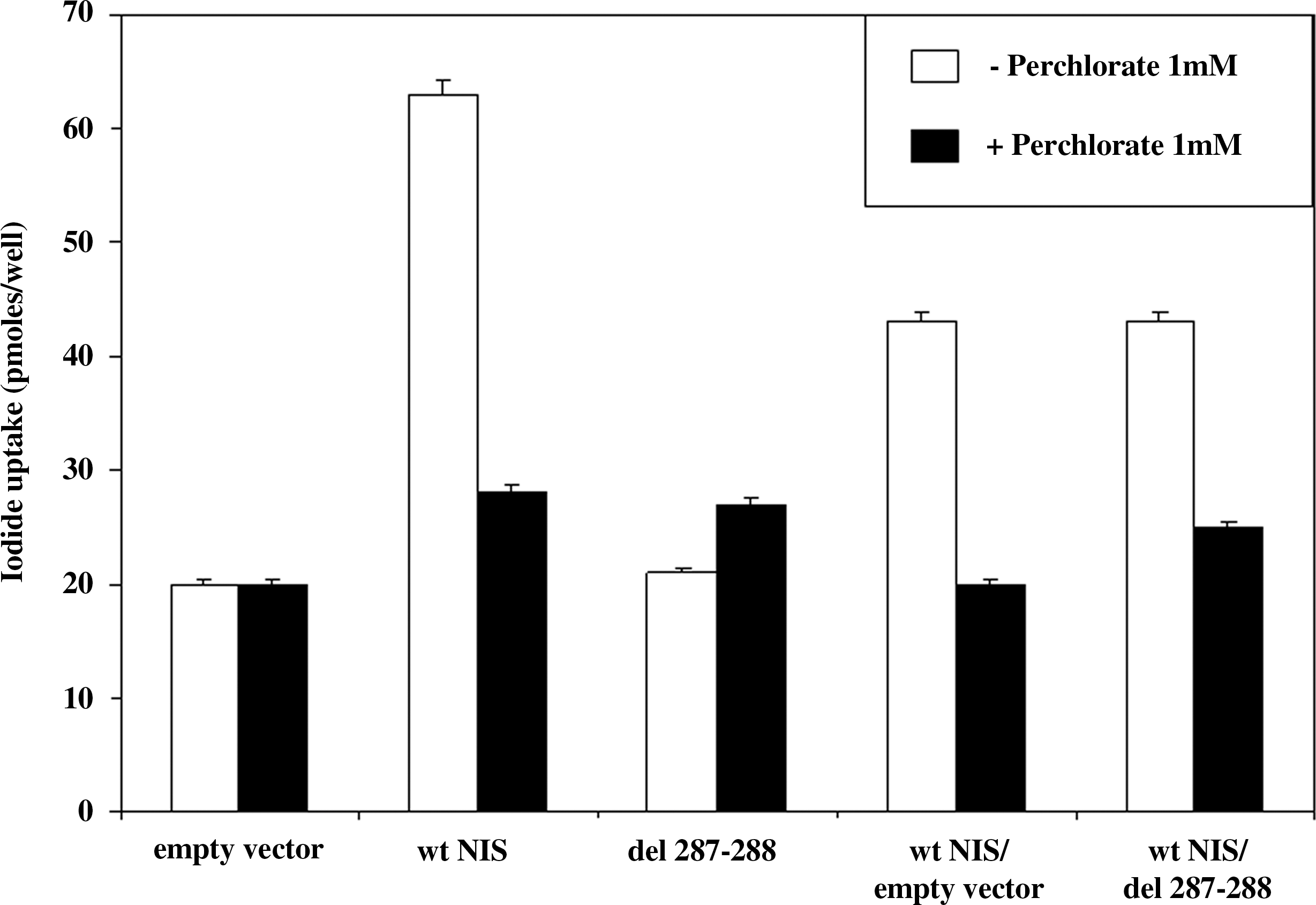
Iodide uptake activity of COS-7 cells transfected with wild-type (wt) hNIS, 1206-1211delGTCGGC, or control empty vector. To represent the heterozygous state of the parents and the brother, COS-7 cells were also transfected with equal amounts of wt NIS and 1206-1211delGTCGGC. Cotransfection of COS-7 cells with wt hNIS and the empty vector was performed as control. In the presence of 1 mM perchlorate, COS-7 cells did not show specific transport of iodide.
Discussion
We identified a novel homozygous deletion in the NIS gene in a patient with CH and features of ITD. The patient was identified at neonatal screening for CH. The parents, who were heterozygous for the same mutation, were said to be unrelated, but some of the ancestors of these two families had lived in the same village many years ago. The diagnosis of ITD was suspected during adolescence on the basis of nodular goiter, hypothyroidism, high serum Tg concentrations (303 ng/mL; normal, <50), and absent uptake of 131I in a normally located thyroid gland and in salivary glands. Direct sequencing of genomic DNA of the proband revealed the presence of a six-nucleotide in-frame deletion, 1206-1211del GTCGGC, of the NIS gene corresponding to the deletion of amino acids 287 and 288 located at the beginning of the VIII transmembrane segment (Figs. 4C and 5). This deletion has not been identified in our control group (200 normal subjects, 400 alleles).
In agreement with the autosomal recessive inheritance pattern of ITD, the patient was homozygous for this deletion, whereas both parents and the brother showed the same deletion in the heterozygous state. The functional study showed that the cells transfected with the mutant 1206-1211del GTCGGC hNIS construct had no perchlorate-sensitive iodide uptake, confirming that the mutation was the direct cause of ITD in this patient. When the mutant was cotransfected with the wt-hNIS, no dominant negative effect was observed. These data are in agreement with the clinical observation that the parents, heterozygous for the mutant allele, have no ITD. Further, the values of the 131I uptake and the capacity to concentrate 131I in the saliva were not affected, suggesting that a defect in iodide uptake is present only in the homozygous condition.
Fifty-five patients with iodide-trapping defects from 29 families have been reported since the first case was published in 1958 (25). The molecular cloning and characterization of NIS has characterized several hNIS gene mutations in patients with ITD (10 –17,19). Although all have an iodide-trapping defect, the clinical features are heterogeneous. Some, like our patient, show severe hypothyroidism, whereas others remain euthyroid without mental or developmental disorders. The inherited pattern of ITD was autosomal recessive in all cases, the heterozygous carriers were always clinically unaffected, and only a few had small goiter. In our patient, hypothyroidism was discovered at neonatal screening. In other cases the age of onset of hypothyroidism, however, was quite variable ranging from the neonatal period to childhood (18). Recently, after reviewing all the clinical cases reported in the literature, Szinnai et al. (18) reported that the age of hypothyroidism onset was genotype specific; they also suggested that the genotype-specific age of hypothyroidism onset may depend on differences in residual NIS protein activity across NIS gene defects. Other environmental factors, however, such as iodide intake may influence the development of hypothyroidism. In fact, a combination of high dietary iodide intake and minimal residual activity due to a 10-fold increase in mutated T354P NIS protein expression has been noted to compensate for the low iodide uptake in a Japanese patient with euthyroid goiter (15).
In our patient her very low thyroid 131I uptake and S/P ratio as measured at the age of 16 were consistent with the neonatal onset of her hypothyroidism. In fact, her 131I uptake values were similar to those described in other patients with ITD identified at neonatal screening.
It seems that the genotype does not influence the thyroid morphological findings that are heterogeneous even in patients carrying the same NIS mutations (18,26). Most patients with ITD have goiter (10 –17,19), and some patients have been reported with follicular adenoma, but a few patients without goiter have been described (15,16,27). Several factors may account for the clinical heterogeneity, including the type of mutation and the patient's iodine intake (14,15). Interestingly, no ITD patients in the Hutterite family described by Kosugi et al. (16) developed goiter. This could be due to a specific characteristic of the mutation or may be explained by their early diagnosis and L-T4 treatment that might have prevented the development of goiter (16). In other cases, however, goiter was observed concomitantly with or years after hypothyroidism despite early hormone replacement therapy (18). It has been proposed that the development of goiter might be due to thyroid growth stimulating effect of TSH, enhanced by low intrathyroidal iodide concentration (28,29). Indeed, extremely high serum TSH levels for a short period of time in early life might greatly enhance the initiation of focal hyperplasia in thyroid follicular cells and result in multinodular goiter at a young age.
In our patient poor compliance with L-T4 treatment, leading to a transient elevation of TSH, was probably a major factor in the induction of her goiter. It is well known that during adolescence compliance to treatment often becomes less regular despite accurate biochemical follow-up and frequent adjustments of therapy (30).
Thyroid scintigraphy is the method of choice for confirmation of the diagnosis of CH and determination of its pathogenesis, but it may be misleading or insufficient to fully characterize the patient's disorder. In fact, there may be no uptake of the isotope, suggesting athyreosis, even though a thyroid gland is present. Thyroid US is useful to determine the presence of an anatomically normal thyroid gland despite lack of imaging by scintigraphy. In our patient a thyroid US was necessary when radioactive iodine scan was unable to detect the thyroid gland to ensure differentiation of thyroid agenesis from a dyshormonogenetic defect due to ITD. In these cases measurement of Tg may also be helpful. In fact, Tg levels are usually normal or even high in patients with CH due to NIS mutations as well as in organification disorders (2).
In patients with a dyshormonogenetic defect, a careful follow-up should be performed particularly during adolescence to avoid a long-term period of inadequate compliance to the treatment, which may favor the development of nodular goiter. In conclusion, we describe a new case of CH caused by a novel deletion in the NIS gene. We also underline the importance of performing a thyroid US when a thyroid scan does not show any iodide uptake in CH patients with elevated Tg values.
Footnotes
Acknowledgments
This work was supported by the following grants. Ministero dell'Università e della Ricerca Scientifica, Programma di Ricerca: Protein, metabolomic, fingerprint, and gene expression profile of thyroid nodules with follicular proliferation cytology—identification of new markers to distinguish benign and malignant thyroid nodules. Programma di Ricerca: Identification and functional study of activating and inactivating mutations and allelic variants of TSH, LH, and FSH receptors. Ministero della Sanità, Ricerca Finalizzata: Indagine sulla associazione fra malattie congenite tiroidee e malattie neuropsichiche rare: studi genetico-molecolari e funzionali.
Disclosure Statement
The authors declare that no competing financial interests exist.