
Abstract
Background:
The multitargeted tyrosine kinase inhibitor sunitinib is known to induce thyroid dysfunction in a substantial proportion of patients treated for advanced renal-cell carcinoma or gastrointestinal stromal tumors. Although sunitinib-induced hypothyroidism seems to be reversible in the majority of patients, some patients develop irreversible thyroid damage resulting in long-lasting thyroid hormone replacement therapy.
Summary:
We report on two cancer patients with a preexisting nodular thyroid gland, who developed thyroid dysfunction and showed marked shrinkage of the thyroid during treatment with the tyrosine kinase inhibitor, necessitating permanent thyroid hormone replacement therapy even after discontinuation of the anticancer agent. Sunitinib treatment in patients with a nodular thyroid can induce a significant decrease in the volume of the enlarged endocrine gland, associated with abnormal thyroid function tests leading to clinical hypothyroidism. The exact pathophysiology remains unknown but we discuss several possible mechanisms of sunitinib-induced thyroid shrinkage.
Conclusion:
Morphological changes of the thyroid gland can be associated with the well-described adverse biochemical effects of treatment with sunitinib and can be a potential marker of the irreversible organ damage.
Introduction
Patient 1
Sorafenib, a TKI, was initiated as the second-line treatment for advanced RCC in a 70-year-old man with a cytokine-refractory RCC. Evaluation of thyroid function at baseline revealed subclinical hyperthyroidism: suppressed thyroid-stimulating hormone (TSH) levels, with normal thyroxine (T4) and triiodothyronine (T3) levels (Table 1). Antithyroglobulin, antithyroid peroxidase, and thyroid-stimulating antibodies were absent. Ultrasound of the thyroid demonstrated a hypoechoic hypervascular solid nodule in the right lobe (largest diameter 28 mm). Due to a computed tomography (CT) scan with contrast 3 weeks earlier, no scintigraphy was performed. Fine-needle aspiration cytology showed a follicular adenoma, thereby excluding metastatic disease to the thyroid. Based on these findings, the nodule was considered as autonomous. Because the peripheral thyroid hormones were normal and the patient had no symptoms or signs of hyperthyroidism, no antithyroid drugs were started. The thyroid volume as measured by CT was 32 mL (1A in Fig. 1). After 13 months of treatment, sorafenib was discontinued due to progressive cancer, and the third-line treatment with sunitinib was installed. TFTs at this time point confirmed subclinical hyperthyroidism (Table 1). The thyroid volume on CT scan measured 22 mL before starting sunitinib at a dose of 50 mg/day in cycles of 4 weeks on and 2 weeks off (1B in Fig. 1). After 9 months, the thyroid gland volume decreased to 13 mL. Ultrasonography showed the largest diameter of the nodule decreased to 20 mm after 5 months of treatment with sunitinib (as compared with 28 mm during sorafenib treatment), while an evolution toward subclinical hypothyroidism occurred (Table 1) with normalization of TSH in the off periods of sunitinib. Together with the occurrence of transient TSH rises, mildly increased serum thyroglobulin levels were observed, suggesting thyroiditis. After 9 months, levothyroxine (LT4) was started because TSH did not recover completely in the off periods. Finally, sunitinib was stopped after 10 months of treatment due to progressive RCC. At that point the thyroid volume assessed by CT was 12 mL (1C in Fig. 1). The patient died due to progressive RCC 4 months thereafter; there was no recovery of thyroid function 2 months after stopping sunitinib, and the dose of LT4 could not be lowered.

Thyroid gland volume reduction on computed tomography scan. Upper images show thyroid gland volume evolution in patient 1. The first image depicts thyroid gland size at the start of tyrosine kinase inhibitor treatment (
Reference values: TSH, normal range: 0.27–4.20 mIU/L; total T4, normal range: 5.1–14.1 μg/dL; FTI, normal range: 4.8–12.7; total T3, normal range: 80–200 ng/dL; Tg, normal range: <25 μg/L.
TSH, thyroid-stimulating hormone; T4, thyroxine; FTI, free thyroxine index; T3, triiodothyronine; Tg, thyroglobulin; ND, not done; LT4, levothyroxine.
In summary, this patient showed a 30% decrease in thyroid volume after 13 months of treatment with sorafenib, and after subsequently being treated with sunitinib for 10 months, again a decrease of almost 50% was noted. There was no change in thyroid function under sorafenib treatment, but development of subclinical hypothyroidism under sunitinib treatment was observed (Fig. 2). This was not reversible at least 2 months after stopping the TKI.
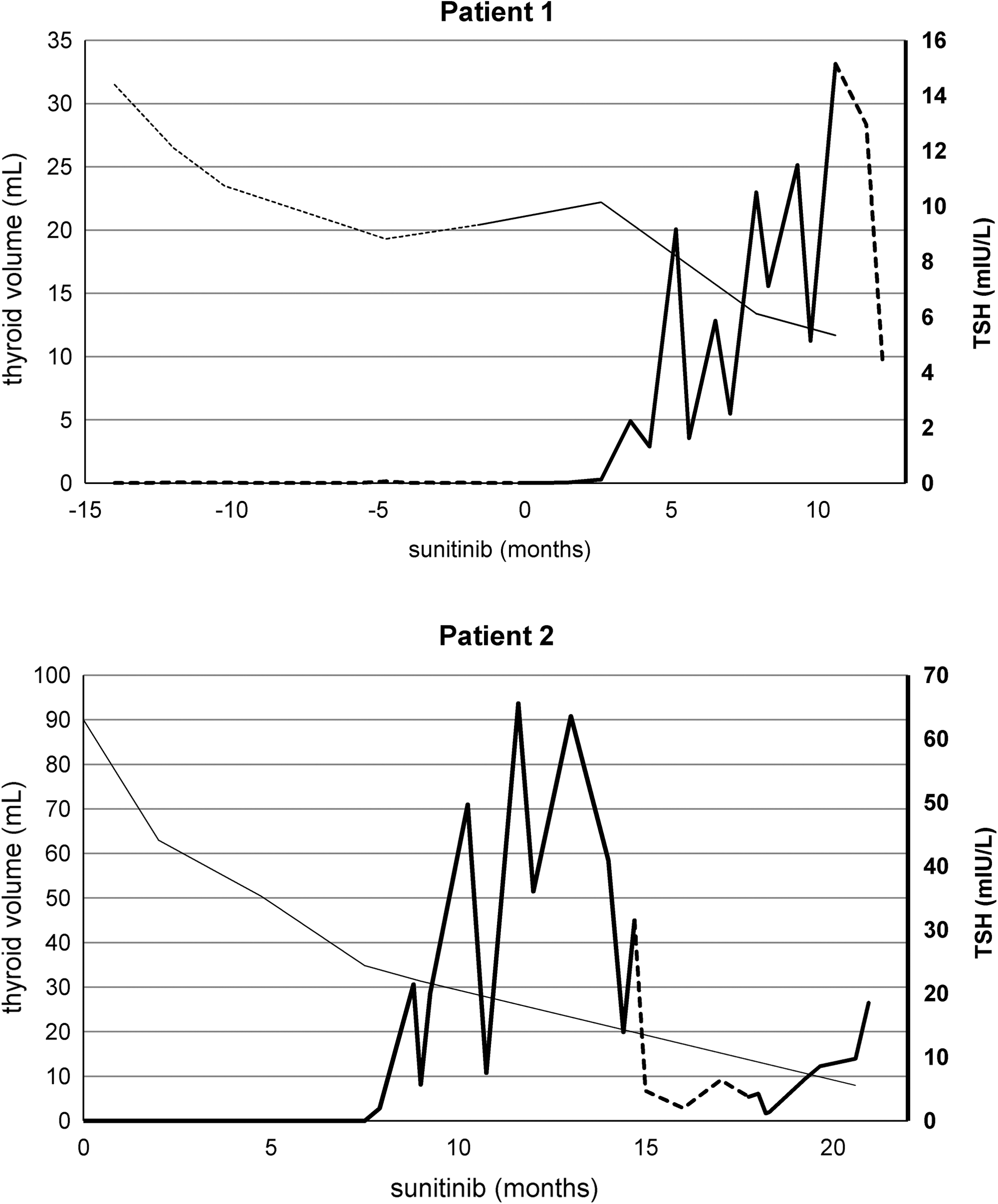
Sunitinib treatment was initiated at 0 months. The thin line demonstrates shrinkage of the size of the thyroid gland during treatment with sunitinib as revealed by volume measurements based on computed tomography imaging (the dotted line in patient 1 delineates sorafenib treatment). Together with thyroid gland size reduction, the bold line shows increased TSH levels during treatment with sunitinib (the dotted line delineates temporary or definitive stop of sunitinib). TSH, thyroid-stimulating hormone.
Patient 2
A 68-year-old man was diagnosed with a multinodular goiter during staging for metastatic RCC by CT scan. On thyroid ultrasound, the nodules were mostly cystic, and the diameter of the largest nodule was 10 mm, and therefore neither suspicious for RCC metastasis nor for primary thyroid malignancy. Therefore, no indication for fine-needle aspiration was present. At first, TFTs revealed a decreased TSH with normal total T4, free thyroxine index, and total T3. Autoantibodies were absent, suggesting the diagnosis of a mild (subclinical) hyperthyroidism due to an autonomous nodular goiter. Thyroglobulin was normal (14 μg/L, normal range <25 μg/L). Due to stable lung and bone metastases, we decided to defer treatment until objective disease progression was present. Six months later, the patient developed intermittent atrial fibrillation and overt iodine-induced hyperthyroidism (Table 1) due to repetitive contrast administration. Treatment was installed with thiamazole 30 mg/day, together with a nonselective beta blocker. On CT the thyroid volume was 90 mL (2A in Fig. 1). Due to progression of RCC, sunitinib was started at a dose of 50 mg/day in cycles of 4 weeks on and 2 weeks off. Two months later, normalization of T4 and T3 occurred, together with highly elevated serum thyroglobulin levels. We considered this together with a clearly changed appearance at ultrasound examination (evolution from isoechoic to clearly hypoechoic) to be very suggestive of destructive thyroiditis. Nine months after initiation of sunitinib, thiamazole was discontinued due to mild TSH elevation (Table 1). The volumetric CT assessment revealed 31 mL for the total gland (2B in Fig. 1). One month later, LT4 was started due to further increase in TSH and decrease of T4 and T3. The patient stopped sunitinib therapy 3 months later after developing cardiac failure but was able to resume the treatment only 4 months later after recovery of cardiac function. Finally, after a total of 21 months with sunitinib treatment, the TKI was stopped due to progressive RCC. At that point TFTs while taking 150 μg LT4 per day revealed mild TSH elevation, and thyroid volume assessed by CT scan confirmed its persistent reduction (2C in Fig. 1). Ten days after stopping the TKI, a recovery of thyroid function could not be observed as the TSH doubled. The patient died 2 weeks later.
In summary, during treatment with sunitinib, the volume of the multinodular goiter decreased 90%, and there was an evolution from subclinical hyperthyroidism (probably through a stage of painless destructive thyroiditis with hyperthyroxinemia because of thyroid hormone leakage) to hypothyroidism (Fig. 2).
Discussion
Sunitinib and other TKIs were initially designed to block specific signaling pathways essential for malignant tumor growth. Although these small molecules are supposed to be quite selective and usually believed to be less toxic as compared to the conventional systemic chemotherapy, they still have some overlap with normal cellular function leading to unpredictable toxicities. TKIs are associated with class-specific or drug-specific adverse events that are not or less frequently seen in patients treated with other systemic antineoplastic agents. With the current wide use of TKIs, the knowledge on the safety profile increases, and it is expected that new or rarer events will emerge in the near future.
Sunitinib-induced thyroid dysfunction, mainly hypothyroidism, is an example of such an unforeseen side effect. Although the effect of the different TKIs on thyroid function varies considerably, thyroid dysfunction most likely represents a class effect of most, if not all, TKIs (8). The incidence of sunitinib-induced thyroid dysfunction is about 60% (8 –12); sorafenib can also cause hypothyroidism, although to a much lesser extent (15). The exact mechanism by which sunitinib causes thyroid dysfunction is unclear.
Several studies have demonstrated that VEGF is expressed in normal, benign, and malignant thyroid tissues (16 –26). Angiogenesis plays an important role in goiter development with endothelial cell proliferation occurring before increased proliferation of the thyroid follicular cells (27). Goiters are usually extremely vascularized, and Viglietto et al. presented data suggesting that placenta growth factor and VEGF, released by thyrocytes in response to the chronic activation of the TSH receptor pathway, may act through a paracrine mechanism on thyroid endothelium thereby inducing goiter hypervascularization (16). Thus, TKI-induced thyroid dysfunction may, at least partially, be due to capillary regression induced by VEGF receptor inhibition (13,14) resulting in a series of steps characterized by the loss of vessel patency, intraluminal fibrin deposition, and cessation of blood flow followed by apoptosis and loss of endothelial cells (14). Capillaries in certain vascular beds seem to exhibit VEGF-dependent plasticity, and the amount of regression was most prominent in the vasculature of the thyroid with a capillary density regression of as much as 68% (13). This mechanism may explain the marked decrease in thyroid size in the aforementioned cases.
Several authors suggest that thyroid hormones themselves may have a proangiogenic effect (28 –30) and that their withdrawal may lead to goiter shrinkage. On the other hand, it is well known that in most patients with sunitinib-induced hypothyroidism, T3 and T4 can be only slightly decreased (11) or can stay within normal range even if TSH is highly elevated. In our two patients, shrinkage of thyroid volume was already observed long before T3 and T4 were occasionally decreased, meaning that thyroid shrinkage cannot solely be explained by the withdrawal of a possible proangiogenic effect of T3 and T4.
Other mechanisms may also play a role. Desai et al. showed evidence of thyroid gland atrophy and mild thyrotoxicosis preceding hypothyroidism, which implies that sunitinib may cause destructive thyroiditis by an unknown mechanism (9). This is consistent with the finding by both Faris et al. and Grossmann et al. that hypothyroidism may be a consequence of sunitinib-induced destructive thyroiditis with transient thyrotoxicosis (31,32). Further, Alexandrescu et al. demonstrated lymphocytic thyroiditis in a patient 7 weeks after starting sunitinib (33). Nonetheless, autoimmunity does not appear to play a major role because Mannavola et al. and Rini et al., respectively, showed that only 1/11 and 5/44 patients developed antithyroid autoantibodies (11,12). Our study supports these data, in that we found none of our 59 prospectively evaluated patients to be having elevated autoantibodies (antithyroglobulin, thyroid-stimulating antibodies, and antithyroid peroxidase), neither before nor after sunitinib treatment, which makes it very unlikely that this destructive thyroiditis is mediated by autoimmunity (8). Mannavola et al. hypothesized that sunitinib-induced hypothyroidism may be the result of impairment of iodine uptake (12), but Salem et al. proved this to be highly improbable because their study demonstrated a slight increase in iodine uptake due to sunitinib (34). Moreover, Wong et al. reported that sunitinib has an antiperoxidase activity of 25–30% compared with that of propylthiouracil (10).
Additional studies are necessary to determine the exact pathophysiologic mechanism of sunitinib-related hypothyroidism and to explain the impressive thyroid volume reduction as observed in the aforementioned patients.
In conclusion, we documented two patients in which sunitinib caused not only hypothyroidism but also a significant reduction in the size of the thyroid gland in previously hyperthyroid patients treated for RCC. Although follow-up after discontinuation of sunitinib in our patients was short, TFTs did not recover completely, suggesting that the described morphological changes of the thyroid might reflect irreversible organ damage. It is important that clinicians treating patients with sunitinib consider the potential side effects of this drug on thyroid function. Sunitinib-induced hypothyroidism may be reversible in the majority of patients; however, marked shrinkage of thyroid volume in sunitinib-treated patients is probably a marker of irreversible thyroid dysfunction necessitating long-lasting thyroid hormone replacement therapy. The underlying molecular mechanisms of sunitinib-induced thyroid dysfunction remain to be elucidated.
Footnotes
Acknowledgments
P.W. is holder of a doctoral fellowship from the Scientific Research Foundation-Flanders (FWO-Vlaanderen). B.D. is holder of a clinical research fellowship (FWO-Vlaanderen).
Disclosure Statement
The authors declare that no competing financial interests exist.