
Abstract
Background:
A thyrotropin (TSH)-secreting pituitary adenoma coexisting with differentiated thyroid carcinoma is rare. There have been only four previously reported cases; three were treated with thyroidectomy followed by pituitary resection and one was treated with thyroidectomy alone.
Methods:
We hereby report the fifth case, in which a patient presented with a TSH/growth-hormone-secreting pituitary macroadenoma coexisting with papillary thyroid carcinoma (PTC).
Results:
She underwent biochemical testing, ophthalmologic examination, thyroid ultrasonography, Tc-99m-pertechnetate thyroid scan, whole-body positron emission tomography, 111In-octreotide scan, thyroid fine-needle aspiration biopsy, octreotide treatment, total thyroidectomy, recombinant human TSH radioactive iodine remnant ablation, and continued treatment with octreotide and levothyroxine after thyroidectomy. She has remained asymptomatic for 24 months without biochemical or radiological evidence of pituitary hormone oversecretion, pituitary adenoma enlargement, and PTC recurrence.
Conclusion:
To our knowledge, this is the first case of a TSH/growth-hormone-secreting pituitary macroadenoma coexisting with PTC being successfully treated with octreotide and levothyroxine after thyroidectomy and recombinant human TSH-stimulated radioactive iodine remnant ablation.
Introduction
Case Presentation
A 57-year-old woman presented with a 4-cm left-sided neck mass of a 4-week duration. She reported 2 months of worsening headache, intermittent nausea and vomiting, fatigue, weakness, palpitations, 20-pound (9.1 kg) weight loss, and mild hand tremors. Her medical history included hypertension, macular degeneration, and uterine fibroids. She had no history of head or neck irradiation and no family history of pituitary or thyroid disorder. Her physical examination revealed an anxious woman with a heart rate of 110 beats/min, a large thyroid goiter with a 6 cm nodule in the left lobe, and fine resting hand tremor. Her laboratory studies showed elevated free thyroxine (FT4) 3.1 ng/dL (0.89–1.76 ng/dL), triiodothyronine (T3) 473 ng/dL (60–181 ng/dL), and elevated TSH 6.1 mIU/L (0.35–5.5 mIU/L). Serum antithyroid peroxidase antibodies, antithyroglobulin antibodies (Tg Ab), thyroid-stimulating immunoglobulins, and thyroxine-binding inhibitory immunoglobulins were all normal. Additional tests showed elevated insulin-like growth factor-1 (IGF-1) 362 ng/mL (92–190 ng/mL) and failure of GH suppression with an oral glucose tolerance test (Table 1). Neck ultrasonography showed a thyroid goiter with a 6 cm left dominant nodule. Magnetic resonance imaging (MRI) of the sella showed a 2.6 cm pituitary mass extending into the left cavernous sinus and encompassing the left internal carotid artery (Fig. 1). Ophthalmologic examination revealed no visual field deficits. Other studies included a Tc-99m-pertechnetate thyroid scan, which revealed intense uptake in the left thyroid nodule. A positron emission tomography whole-body survey showed no evidence of metastasis. An 111In-octreotide scan demonstrated increased uptake in the pituitary mass and thyroid nodule (Fig. 2).

Pituitary magnetic resonance imaging at presentation showed a 2.6 × 2.4 × 1.5 cm pituitary adenoma extending superiorly to the optic chiasm and laterally into the left cavernous sinus and encompassing the left internal carotid artery.

111In-octreotide scan showed increased uptake in the pituitary gland and left thyroid lobe, corresponding to the known pituitary tumor and left thyroid nodule.
TSH, thyrotropin; FT4, free thyroxine; T3, triiodothyronine; IGF, insulin-like growth factor; Tg Ab, antithyroglobulin antibodies; LAR, long acting release; SQ, subcutaneous; rhTSH, recombinant human TSH.
She was started on short-acting octreotide and transitioned to long-acting octreotide with a plan for transsphenoidal sinus surgery (TSS) 6 weeks after initiation of the octreotide therapy. She also underwent fine-needle aspiration of the left thyroid nodule, whose cytology showed follicular neoplasm. We recommended that she undergo thyroidectomy after TSS. However, she subsequently declined TSS and opted to continue medical treatment with octreotide. Six months after her initial presentation, while remaining on octreotide, she underwent total thyroidectomy with central lymph node evaluation. The surgery was uneventful and postoperative course was without any complications. The weight of her thyroid gland was 42 g. The right lobe measured 4.5 × 2.0 × 1.0 cm and the left lobe measured 5.5 × 3.5 × 2.7 cm. The histopathology showed multifocal PTC, with one focus in the left lobe and 2 foci in the right lobe; the largest focus was 0.8 cm within the left thyroid mass. Due to the concern for the TSH-oma enlargement, thyroxine withdrawal was considered inappropriate. She was started on levothyroxine suppression immediately after the thyroidectomy and subsequently underwent rhTSH-stimulated RAI remnant ablation with 100 mCi of 131I and continued to be treated with both octreotide and levothyroxine. The postablation 131I scan showed uptake only in the thyroid bed, compatible with the postthyroidectomy status. The baseline serum Tg level at presentation was greater than 3000 ng/mL and Tg Ab was less than 20 IU/mL (Quest Laboratory, Chantilly, VA). One month after thyroidectomy, her Tg decreased to 0.3 ng/mL and Tg Ab remained less than 20 IU/mL.
Her serum TSH, FT4, and T3 normalized 4 days after initiating octreotide therapy, whereas GH normalized after 1 month. All her symptoms resolved 2 months after the initiation of octreotide; repeat pituitary MRI at that time showed significant tumor shrinkage. She has remained asymptomatic without evidence of recurrent pituitary hormone oversecretion or pituitary tumor enlargement. At 20-month follow-up laboratory results revealed serum IGF-1 171 ng/mL, GH 0.7 ng/mL, FT4 1.6 ng/dL, and TSH 0.28 mIU/L, demonstrating biochemical control of TSH and GH secretion from the pituitary adenoma. One year after thyroidectomy and RAI remnant ablation, PTC surveillance under rhTSH stimulation showed undetectable Tg (<0.1 ng/mL) and Tg Ab (<0.4 IU/mL) (USC Laboratory, Los Angeles, CA) as well as negative 123I scan (Fig. 3) with TSH level of 46 mIU/L, indicating remission of the PTC.

Recombinant human thyrotropin-stimulated 123I scan at 1-year follow-up.
Discussion
Coexistence of a TSH-oma and differentiated thyroid carcinoma is rare. Review of the literature showed only four reported cases of TSH-omas with coexisting thyroid carcinoma. These patients presented with a thyroid nodule, goiter, or hyperthyroidism (1 –4).
When a patient presents with elevated thyroid hormones and inappropriately elevated TSH, TSH-omas should be suspected. However, the differential diagnosis for inappropriate TSH secretion should include resistance to thyroid hormone (5 –8). This condition is an autosomal dominant disorder characterized by a mutation in the thyroid hormone beta receptor gene, which leads to failure of the pituitary to detect peripheral thyroid hormones, resulting in increased TSH secretion. The two diagnoses can be differentiated with TSH-releasing hormone (TRH) stimulation test, T3 suppression test, molar ratio of alpha subunit/TSH, and pituitary MRI. In patients with TSH-omas, TSH usually remains unchanged in response to TRH stimulation as well as T3 suppression tests due to autonomy of the TSH-oma. Patients with resistance to thyroid hormone, however, tend to show increased TSH with TRH stimulation and decreased TSH with T3 suppression due to preserved responsiveness of the normal thyrotrophs. In a hypergonadotropic postmenopausal patient with elevated TSH, an alpha subunit/TSH ratio greater than 1 would be suggestive of a TSH-oma (7,9). Our patient had an alpha subunit/TSH ratio of 6. Neither TRH stimulation nor T3 suppression test was performed due to the unavailability of TRH and the hyperthyroid state of the patient. With inappropriately elevated TSH, high FT4, T3, alpha subunit, and alpha subunit/TSH molar ratio in the presence of a pituitary adenoma and the absence of family history of thyroid disorder, we were able to confidently exclude thyroid hormone resistance.
After diagnosing her TSH-oma, we also tested her other pituitary hormones, which showed cosecretion of GH as demonstrated by elevated IGF-1 levels and failure of GH suppression with oral glucose challenge test. Despite laboratory evidence of GH oversecretion, she did not exhibit overt physical manifestations of acromegaly. We speculated that lack of acromegalic features might have been due to early detection of her pituitary tumor and mild GH oversecretion. Nevertheless, the exact duration of her TSH-oma is unknown despite her normal TSH profiles in 2001 and 2004 as normal TSH alone without concurrent FT4 and T3 values cannot reliably exclude TSH-omas (5).
Once a TSH-oma is diagnosed, there are several treatment options. While surgical resection is the treatment of choice in most cases, it only cures about one third and improves tumor size and symptoms in another third (5,7). Medical management with either a somatostatin analog or a dopamine agonist has been reported (10 –14). Two previous publications (15,16) have shown that TSH/GH-omas can be successfully treated with octreotide. The first case showed both biochemical improvement and tumor shrinkage (15), whereas the second case showed clinical and biochemical improvement without tumor size reduction (16). Octreotide scintigraphy can be performed before somatostatin analog treatment; however, Socin et al. (17) did not find a strong predictive correlation between positive octreotide scans and TSH-oma responsiveness to this therapy. In this patient, we had recommended TSS as the primary treatment, which she declined. Subsequently, we started her on short-acting octreotide and then transitioned to long-acting octreotide. Similar to a previously reported case in which a TSH/GH-oma had undergone significant volume reduction with short-term octreotide therapy (15), our patient's tumor volume decreased by 50% after 6 weeks of octreotide treatment.
As there have been several reported cases of a TSH-oma coexisting with differentiated thyroid cancer (1 –4), we believe a patient with a TSH-oma should be screened for possible thyroid nodules with thyroid ultrasonography. When a patient is found to have a TSH-oma coexisting with differentiated thyroid carcinoma, the treatment sequence for these tumors has not been well established. Ohta et al. (4) suggests that in this situation, thyroidectomy should be carried out before adenomectomy even though this order of treatment may result in TSH-oma progression due to failure of TSH inhibition. On the contrary, we believe a TSH-oma should be treated before thyroidectomy, as TSH has been suggested to be the stimulus for thyroid tumor growth (1 –3). In addition, when a TSH-oma and thyroid goiter or nodules coexist, we favor total thyroidectomy independent of fine needle aspiration cytology results, as there is a potential for the goiter, nodule, or hyperthyroidism to recur in the remaining thyroid tissues (12).
Beck-Peccoz et al. (5) suggested that autonomous thyrotrophs may respond to a small decrease in thyroid hormones by hyperproliferation, developing into macroadenomas. Therefore, we started our patient on levothyroxine immediately postoperatively. Six weeks after thyroidectomy, she received RAI remnant ablation under rhTSH stimulation (18,19) instead of the more traditional thyroid hormone withdrawal, which could prolong the hypothyroid state and promote adenoma growth due to lack of negative feedback inhibition. In addition, we chose to continue her octreotide treatment, as a TSH-oma can re-grow after octreotide discontinuation (12). She has done well on the combination therapy with octreotide and levothyroxine. Her adenoma volume decreased by 50% at 2-month follow-up and has remained stable for the past 20 months (Fig. 4). Like Socin et al. (17), we believe that lack of complete tumor shrinkage is likely due to the fibrotic nature of these TSH-omas. Given her diagnosis of multifocal PTC, we plan to maintain her levothyroxine at a dose to achieve slight TSH suppression. Interestingly, her serum TSH level has remained mildly suppressed in the presence of normal FT4, suggesting that octreotide possibly inhibits TSH secretion from both tumoral and normal thyrotrophs, consistent with the observation of Mannavola et al. (20).
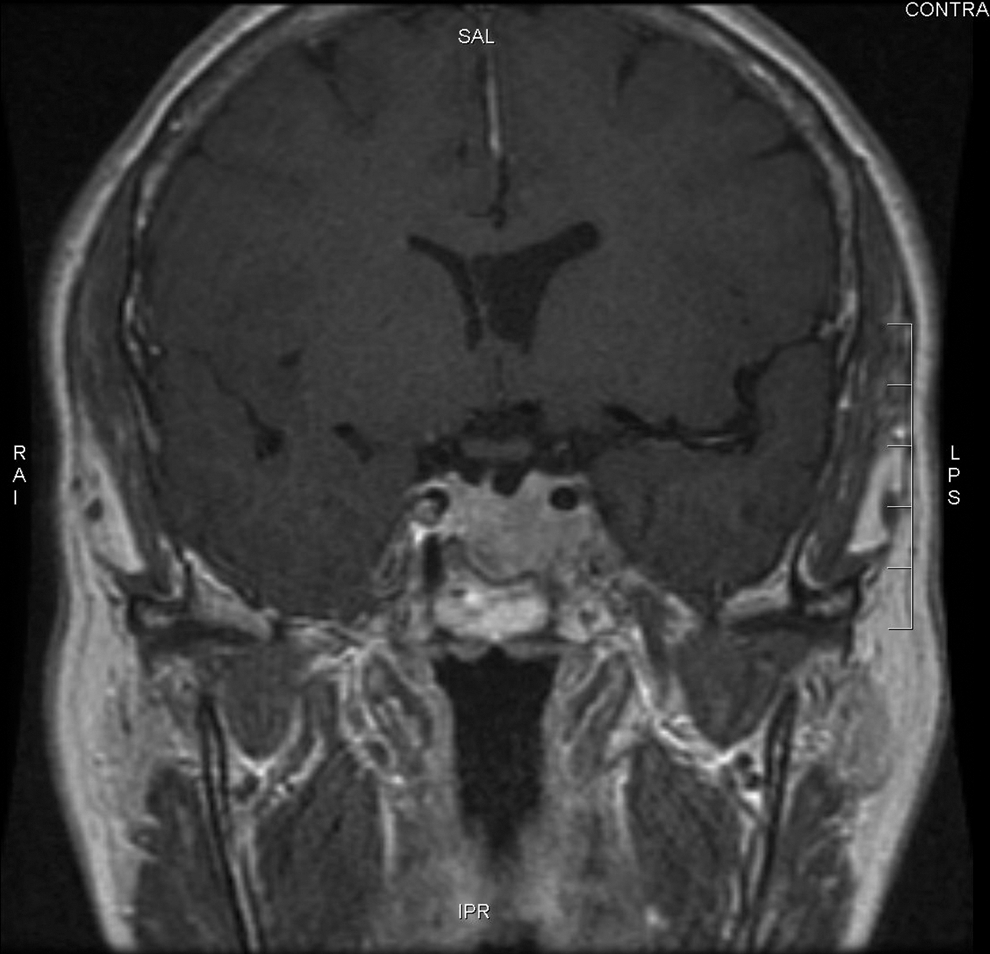
Pituitary magnetic resonance imaging at 20-month follow-up showed a 1.3 × 2.4 × 1.6 cm pituitary adenoma with persistent extension into the left cavernous sinus. Mass effect on optic chiasm has lessened.
To our knowledge, this is the first case in which a TSH/GH-oma coexisting with PTC was successfully treated with long-acting octreotide and levothyroxine after thyroidectomy and rhTSH-stimulated RAI remnant ablation. Whether this treatment modality will lead to prolonged stabilization of the TSH/GH-oma remains to be established. Controlled trials with long-term follow-up will be necessary to determine whether thyroidectomy followed by combined medical therapy with levothyroxine and a somatostatin analog may offer another effective alternative to pituitary resection in selected patients with TSH-omas who decline or are not surgical candidates for TSS.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the U.S. Government.
Disclosure Statement
The authors declare that no competing financial interests exist.