
Abstract
Objective:
Thyroid cancer, the commonest of endocrine malignancies, continues to increase in incidence with over 19,000 new cases diagnosed in the European Union per year. Although nonmedullary thyroid cancer (NMTC) is mostly sporadic, evidence for a familial form, which is not associated with other Mendelian cancer syndromes (e.g., familial adenomatous polyposis and Cowden's syndrome), is well documented and thought to cause more aggressive disease. Just over a decade ago, the search for a genetic susceptibility locus for familial NMTC (FNMTC) began. This review details the genetic studies conducted thus far in the search for potential genes for FNMTC.
Design:
An electronic PubMed search was performed from the English literature for genetics of FNMTC and genetics of familial papillary thyroid carcinoma (subdivision of FNMTC). The references from the selected papers were reviewed to identify further studies not found in the original search criteria.
Main Outcome:
Six potential regions for harboring an FNMTC gene have been identified: MNG1 (14q32), TCO (19p13.2), fPTC/PRN (1q21), NMTC1 (2q21), FTEN (8p23.1–p22), and the telomere-telomerase complex. Important genes reported to have been excluded are RET, TRK, MET, APC, PTEN, and TSHR.
Conclusion:
The genetics of FNMTC is an exciting field in medical research that has the potential to permit individualized management of thyroid cancer. Studies thus far have been on small family groups using varying criteria for the diagnosis of FNMTC. Results have been contradictory and further large-scale genetic studies utilizing emerging molecular screening tests are warranted to elucidate the underlying genetic basis of FNMTC.
Introduction
Familial NMTC (FNMTC) is often categorized into two groups. The first group accounts for a minority of cases and is called syndromic FNMTC. This group is characterized by a preponderance of nonthyroidal tumors and is associated with Mendelian cancer syndromes such as familial adenomatous polyposis and Cowden's syndrome. The second group, nonsyndromic FNMTC, is characterized by a preponderance of NMTC. While genetic loci for syndromic FNMTC have been identified and well documented, the genetics of nonsyndromic FNMTC remains ambiguous. The aggressiveness of FNMTC compared with its sporadic counterpart has also been reported by a number of studies. Evidence of a younger age at presentation with higher incidence of tumor multifocality, local invasion, lymph node metastasis, and local or regional recurrences have fuelled the drive to locate a susceptibility gene (4 –6,14 –17). In addition, a recent study has found FNMTC to display features of clinical anticipation where individuals in the second generation appear to have more advanced disease at presentation with an earlier age of onset (17). Some groups suggest that these patients should be managed more aggressively than those with sporadic NMTC. Recommendations vary but include offering total thyroidectomy with prophylactic central neck lymph node dissection as the first operation, followed by postoperative radioiodine ablation, and thyroid hormone suppression regardless of the primary tumor size (6,17 –20). Although prophylactic thyroidectomy in FNMTC has been suggested, this recommendation remains debated (18). At present, there are no reliable screening tools for FNMTC. Identification of molecular markers would permit screening, stratification of management, and potentially treatment of patients at high risk before disease development.
Molecular techniques
With recent technical advances in molecular genetics, a number of potential loci for an FNMTC gene have been identified. To evaluate the significance of these reports, a basic understanding of the genetic methods is required.
With the completion of the Human Genome Project, there now exists an extensive map of molecular markers throughout the entire genome. The most commonly used molecular markers are microsatellites (polymorphic loci with a variable number of tandem repeats) or single-nucleotide polymorphisms in which the relevant DNA sequence varies only by a single base. By analyzing the distribution of these markers in individuals with disease, a marker may be discovered that frequently segregates with the disease, hence exhibiting linkage. Genetic linkage is defined as the tendency for alleles that are close together on the same chromosome to be inherited together as an intact unit (Fig. 1). A mathematical calculation is used to calculate the log 10 of odds (LOD score) to support or refute linkage. An LOD score of 3 or more is accepted as confirmation of linkage and a score of −2 or less is indicative of nonlinkage. Typically, an initial two-point linkage analysis is roughly performed to map the disease locus to a chromosome region. This is often followed by multipoint linkage analysis, which narrows down the region where the disease gene may be located using markers that are specifically known to that region (Fig. 2). The centiMorgan (cM) is the unit of measurement for genetic distance. If two loci are 1 cM apart, then a recombination event will occur between them in 1% of cases, that is, they will cosegregate in 99% of cases. This is different from physical distance, which is measured in base pairs. One centiMorgan corresponds to approximately 1 million base pairs or 1 Mb.
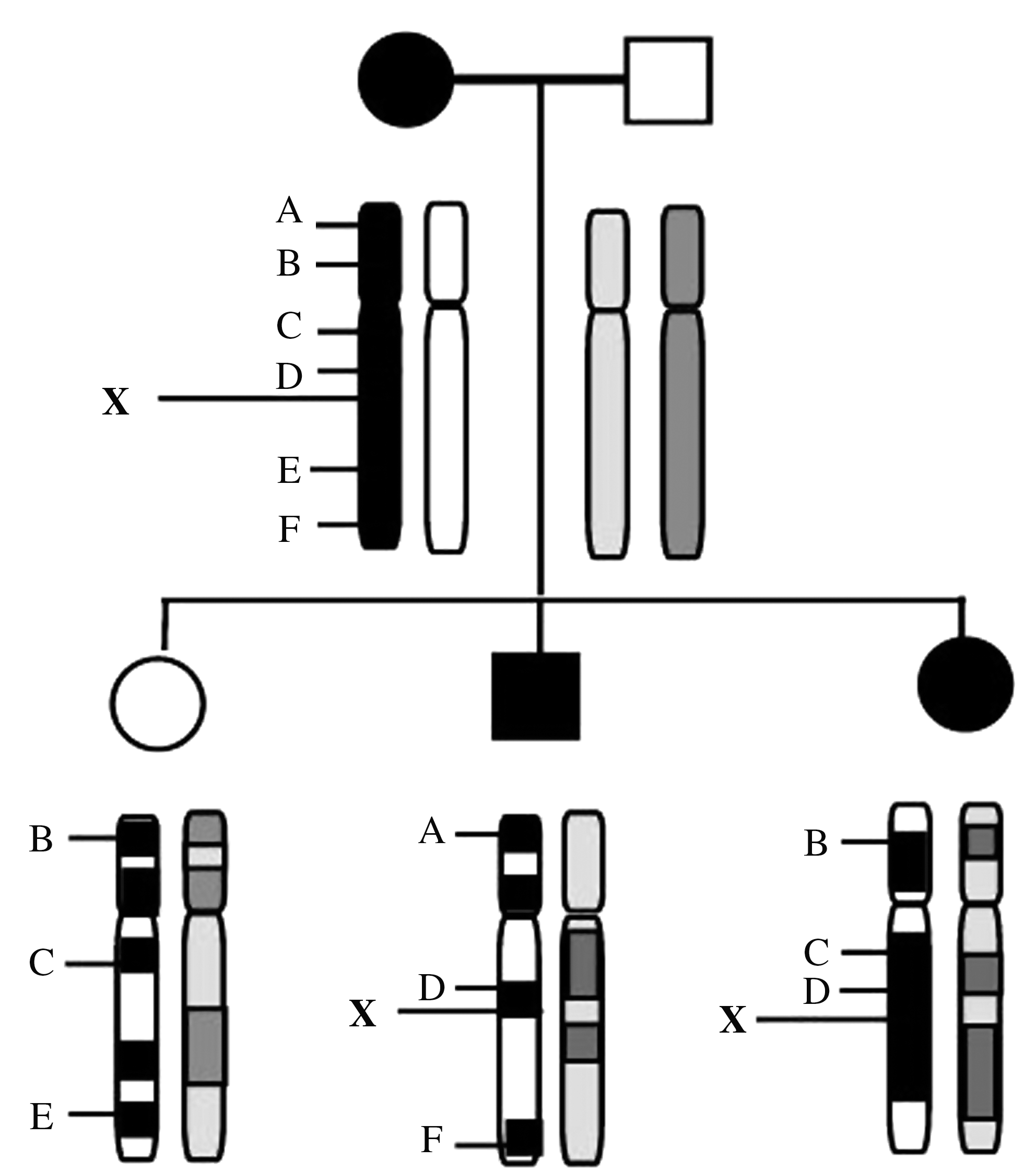
Linkage analysis. The unknown disease gene is represented by X. A–F are genetic markers whose exact chromosomal positions are known. If we find that a particular marker always segregates with the disease (in this case marker D), the two loci may be linked, that is, they are so close together that they are unlikely to be separated by crossover.
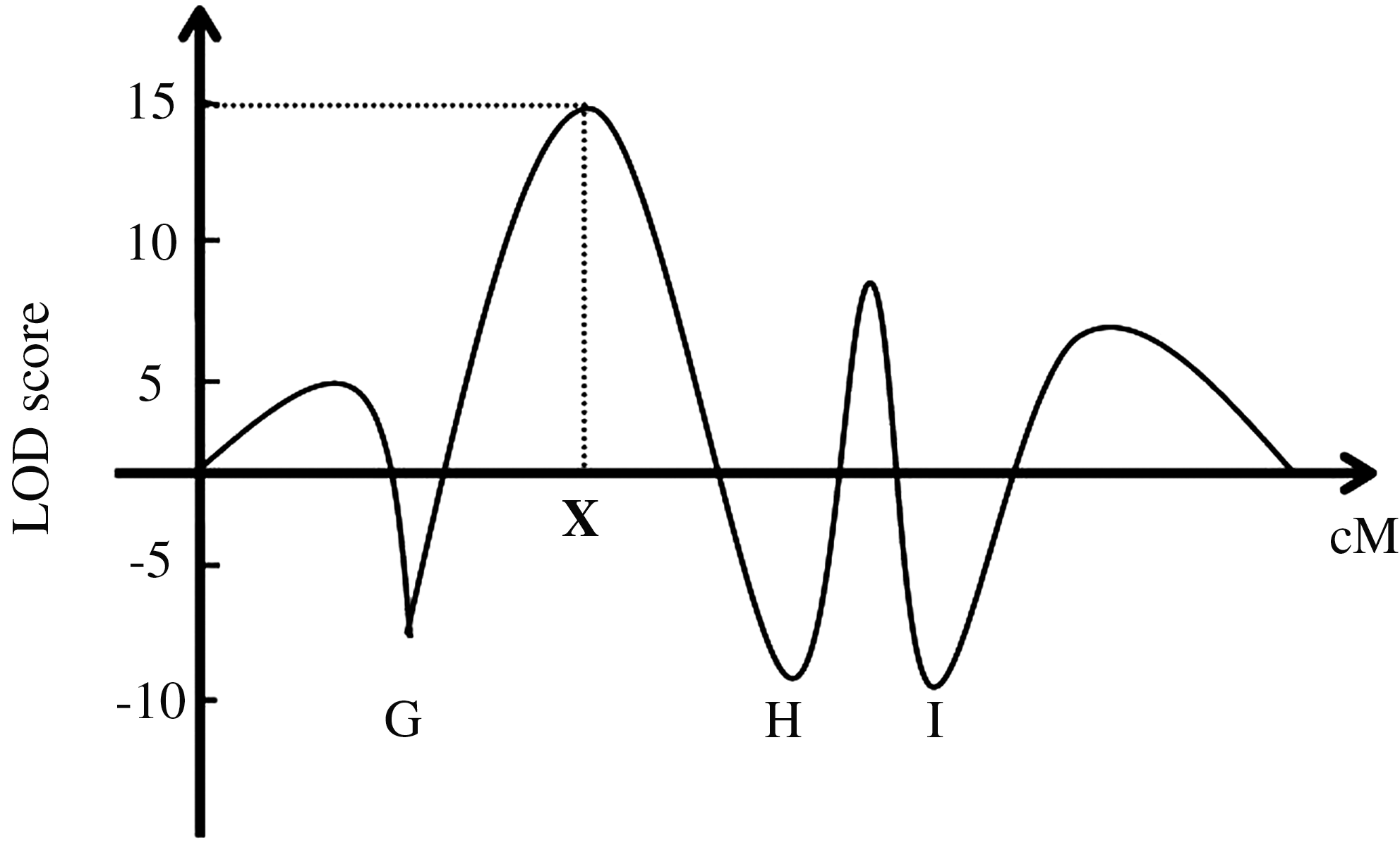
Multipoint linkage map. G–I are genetic markers whose position on the genetic map are known. The highest LOD score in this case is +15, and it indicates the most likely position of the disease gene.
Linkage disequilibrium describes the occurrence of two or more alleles at linked loci more frequently than expected by chance. The demonstration of linkage disequilibrium can be very useful when trying to map a disease gene, as it implies that the marker locus is in very close proximity to the disease gene; hence, recombination events are less likely to have occurred.
A limiting factor in linkage analysis is locus heterogeneity. This is where a disease phenotype can be caused by mutation at different chromosome loci. As a result, some studies calculate the heterogeneity of LOD (HLOD) score rather than the homogeneity of LOD (LOD) score (21).
Loss of heterozygosity (LOH) is another important finding that aids identification of the disease gene. This is where a cell carrying a mutated allele in a tumor suppressor gene loses the corresponding normal allele, allowing the germline mutation to be unmasked. Known genes in the area of LOH can then be examined and mapped.
Basic chromosome nomenclature is used to describe the position of a particular genetic locus. Each chromosome has a short (p) and a long (q) arm. Each arm is then subdivided into bands. For example, the locus 12p11 would indicate that the locus is on band 11 on the short arm of chromosome 12.
Studies employing these molecular techniques have revealed potential loci and also excluded some important genes that were thought to be good candidates in susceptibility to FNMTC. This review examines genetic studies of nonsyndromic FNMTC that have been published to date.
Method
An electronic PubMed search was performed from the English literature for genetics of FNMTC and genetics of familial papillary thyroid carcinoma (subdivision of FNMTC). References from the selected papers were reviewed to identify further studies not found in the original search criteria.
Putative regions of genetic susceptibility
MNG1 14q31 locus (MIM 138800)
In 1997, Bignell et al. published a potential susceptibility locus for FNMTC. A large Canadian pedigree with 18 cases of multinodular goiter (MNG) and 2 cases of papillary carcinoma was studied. Their rationale for studying patients with MNG while trying to elucidate a susceptibility locus for FNMTC was the observation that benign thyroid disease is a common occurrence in NMTC (22). On genotyping 34 individuals, a potential susceptibility locus at 14q31 was identified. A maximum two-point LOD score of 3.8 and a multipoint LOD score of 4.88 were achieved. Haplotype analysis concurred with the linkage data and showed an autosomal dominant mode of inheritance. To confirm these initial findings, linkage analysis was repeated on 37 smaller pedigrees, each containing at lease two cases of NMTC. However, this failed to show evidence of linkage and produced LOD scores of −8.84 (22).
Five further studies failed to find linkage between the MNG1 locus and FNMTC (see Table 1). The largest of these studies genotyped 150 individuals from 56 pedigrees containing 128 NMTCs. This showed no evidence of linkage to 14q31 (23). McKay et al. also looked at FNMTC in a large Tasmanian family of 33 members (24). Despite not finding evidence of linkage to MNG1, the study observed a much higher penetrance of papillary thyroid carcinoma (PTC) when compared with the Canadian family. It was suggested that given there are different phenotypes of families with FNMTC, there may well be different genes for each phenotype.
FNMTC, familial nonmedullary thyroid cancer; MNG, multinodular goiter; TCO, thyroid tumors with cell oxyphilia; PRN, papillary renal neoplasm; LOH, loss of heterozygosity; HLOD, heterogeneity of LOD; LOD, log 10 of odds; PTC, papillary thyroid carcinoma; FPTC, familial papillary thyroid carcinoma.
This idea was supported by Malchoff et al., who performed linkage analysis on a three-generation kindred in the United States with five members affected with PTC and two with renal-cell carcinoma. In this phenotype, a novel candidate locus was identified at 1q21 [see fPTC/PRN 1q21 locus (MIM 605642) section] but linkage to the MNG1 locus was not found (25). Only women over the age of 30 were used to calculate LOD scores in this study. Men and individuals less than 30 years were not used as accurate figures, as penetrance was not available. Given that FNMTC is thought to occur in younger age groups and more commonly in men than its sporadic counterparts (17), this exclusion may have diluted the results. A subsequent study looking for linkage to MNG1 involved genotypic analysis of seven families consisting of 20 patients with FNMTC. FNMTC was diagnosed in this study using a criterion of three or more affected first-degree relatives (including benign thyroid disease). Once again, linkage analysis failed to show any association with the MNG1 locus (26).
In summary, the MNG1 locus has only shown evidence of linkage to FNMTC in one Canadian kindred with multiple MNGs. Linkage analyses in a further 124 families have failed to confirm an association between MNG1 and FNMTC, suggesting that this locus may not be involved in FNMTC or that it may account for only a minority of FNMTC cases with MNG. Alternatively, it is possible that the MNG1 locus may harbor a gene for MNG alone but not for FNMTC.
TCO 19p13.2 (MIM 603386)
The thyroid tumors with cell oxyphilia (TCO) locus were first mapped in a three-generation French family consisting of six cases of MNG and three of PTC (27). Linkage analysis followed by physical mapping found a locus on chromosome 19p13.2. Both benign and malignant thyroid tumors in this family were noted to have cell oxyphilia (27). It was speculated that the TCO locus is associated only with this unique form of FNMTC with cell oxyphilia. However, when Bevan et al. performed linkage analysis on 22 FNMTC families, they found 1 family with linkage to the TCO locus and, interestingly, the thyroid cancers in this family did not display cell oxyphilia (28).
McKay et al. genotyped an additional eight FNMTC families with tumor cell oxyphilia and found linkage to TCO but with a low LOD score at 1.56 (29). This study suggested that the TCO and NMTC1 loci (another potential genetic locus) may interact to increase the risk of FNMTC [see NMTC1 2q21 locus (MIM 606240) section].
Stankov et al. found LOH at the TCO site in 70 sporadic oxyphilic thyroid tumors, but these numbers were too small to draw statistically significant conclusions (30). Stankov et al. also suggested that TCO might be a tumor suppressor gene. This was supported by a smaller study by Prazeres et al. that assessed LOH in 14 cases of FNMTC in nine families (31). In three of these families, a nonrandom retention of the TCO haplotype suggested selective loss of the normal allele. Again, this could be the result of inactivation of a tumor suppressor gene. Due to the small numbers, linkage could not be accurately assessed, but 8 out of 14 cases showed LOH at 19p13.2.
Since the original mapping of TCO, 4 subsequent studies have shown no association to this locus in FNMTC in a total of 65 kindreds (see Table 1).
In summary, the TCO locus may account for FNMTC in a minority of cases, most of which are associated with tumor cell oxyphilia. This is a rare type of thyroid cancer with distinct morphology and even in sporadic cases of thyroid cancer with cell oxyphilia, an association with the TCO locus has been noted.
fPTC/PRN 1q21 locus (MIM 605642)
Malchoff et al. described a large three-tiered kindred with five cases of PTC and two of papillary renal neoplasm (PRN) (25). Thirty-one members of this family were genotyped. A locus on chromosome 1q21 was identified to give a maximum 3-point LOD score of 3.58. Haplotype analysis revealed that all affected subjects carried the same phenotype within the region of linkage. To date, no further families with a PTC and PRN association have been reported. Two studies that performed linkage analysis on a total of 29 FNMTC families (without PRN) did not find an association between FNMTC and the fPTC/PRN locus (see Table 1). These findings suggest that the fPTC/PRN locus may harbor a susceptibility gene for a unique FNMTC phenotype where PTC is associated with PRN. However, this is a very rare phenotype, and no association to this locus is suggested for the vast majority of FNMTC.
NMTC1 2q21 locus (MIM 606240)
McKay et al. reported another potential susceptibility locus for FNMTC, this time in a large Tasmanian pedigree with numerous PTCs (32). An extensive genome-wide scan followed by haplotype analysis revealed that seven out of eight subjects with PTC shared a common haplotype on chromosome 2q21. To confirm the findings, linkage analysis was performed on an additional 80 pedigrees with FNMTC. This showed significant linkage to the 2q21 locus with a multipoint HLOD score of 3.07. When these families were stratified on the basis of containing at least one case of follicular variant of PTC (fvPTC), the HLOD score increased to 4.17, suggesting that the 2q21 locus has a more significant association with fvPTC. Tumors with cell oxyphilia were excluded.
McKay et al. performed linkage analysis on a further 10 FNMTC families, 9 of which contained thyroid cancers with cell oxyphilia. This showed evidence for linkage of FNMTC to the NMTC1 locus with an LOD score of 2.85. The authors then went on to perform a two locus linkage analysis to explore the possibility of an interaction between the NMTC1 and TCO loci. This revealed significant evidence in favor of a two locus inheritance model with an LOD score of 3.92, suggesting that interaction between the TCO and NMTC1 loci may increase the risk of FNMTC in patients who inherit both susceptibility loci (29). However, the results appear to be skewed by a large Tyrolean family who alone achieved a two locus LOD score of 3.21. Evidence for interaction between the TCO and NMTC1 loci was further assessed by Stankov et al. (30), who reanalyzed the French family on whom the TCO1 locus was originally mapped. No evidence of linkage to the NMTC1 locus was found. This questions the digenic inheritance hypothesis suggested by McKay et al. (29), but an alternative explanation could be that the two loci present an individual risk in different phenotypes of FNMTC. However, when present together (which may be rare), the risk of FNMTC is significantly increased.
Another small study looked at LOH at the NMTC1 locus in FNMTC and showed that 2 out of 14 cases of FNMTC exhibited LOH at the NMTC1 locus (31). This study was too small to allow linkage analysis to be performed. A later study of seven families found no evidence of linkage to the NMTC1 locus (26).
In summary, few studies have looked at the association between FNMTC and the NMTC1 locus. The largest of these studies suggests that fvPTC has a more significant association to the NMTC1 locus than familial papillary thyroid carcinoma (FPTC). The NMTC locus is also associated with some oxyphilic tumors.
Telomere-telomerase complex
One study has examined the telomere-telomerase complex. Telomeres are specialized DNA sequences found at the end of each chromosome arm acting as a cap to maintain genetic/structural integrity. During cell division, telomere length is maintained by the reverse transcriptase enzyme, telomerase. Without this enzyme, telomeres would progressively become shorter with every cell division until a critical length is reached when the cell would no longer divide. Telomerase activity is usually suppressed in somatic cells, allowing them to age and replicate only a finite number of times. However, in 80–85% of cancer cells increased telomerase activity facilitates prolonged cell survival. This observation prompted Capezzone et al. to study the telomere-telomerase complex in 47 patients with FNMTC (33). Analysis revealed significantly shorter telomere lengths, higher telomerase reverse transcriptase (hTERT) gene amplification, and hTERT mRNA expression in patients with FPTC when compared with sporadic PTCs. However, the study did not report any mutations of the hTERT gene or the telomerase RNA component. This may suggest that the increased telomerase activity is caused by a gene mutation that directly up-regulates telomerase gene expression.
As yet, no other groups have assessed the association between the telomere-teromerase complex and FNMTC. The exact mechanism of a potential association remains unclear.
FTEN 8p23.1–p22
Cavaco et al. performed linkage analysis using single-nucleotide polymorphisms followed by microsatellites on a family with 11 cases of benign thyroid disease and 5 cases of thyroid cancer (34). Linkage was confirmed to the 8q23.1–p22 locus with a maximum LOD score of 4.41. However, linkage analysis on a further six families showed LOD scores of −2 or lower for this locus. Further studies are warranted before this locus is considered to harbor an FNMTC gene.
Important genes excluded in FNMTC
While several studies have suggested putative FNMTC loci, some important genes have been excluded as the cause of FNMTC.
The adenomatous polyposis coli (APC) gene associated with familial adenomatous polyposis and the phosphate and tensin homolog (PTEN) gene associated with Cowden's syndrome are both involved in syndromic FNMTC (34). However, no association has been seen with nonsyndromic FNMTC (24,25,27–28). Mutations of the thyroid-stimulating hormone receptor locus have been found in sporadic thyroid adenomas, making this a potential site for an FNMTC gene (27–28,35), but three independent studies have failed to show an association (24,27–28). Mutations of the rearranged during transfection (RET) gene are present in a large proportion of medullary thyroid cancers and this gene is often rearranged in sporadic NMTCs (9,35–36), but four studies have found no association with FNMTC (24,25,27,36). Tropomyosin receptor kinase and MET (a hepatocyte growth factor receptor) are also oncogenes found to be altered in sporadic NMTC but not in FNMTC (24,25,27). The TRKA gene, located close to the fPTC/PRN locus, failed to show an association with FNMTC (36). JUNB (proto onco–gene) was thought to be a promising site for an FNMTC gene, as it is known to be an oncogene and is located at the TCO locus (28); however, no association with FNMTC has been found (37).
The V–raf murine sarcoma viral oncogene homolog B1 (BRAF) locus has a high prevalence of mutations in PTC, in particular the T1799A BRAF mutation (38). After direct DNA sequencing of 26 individuals, one study found no T1799A BRAF mutation (38). However a second study by Cavaco et al. did find somatic BRAF mutations in 14 out of 27 patients with FNMTC (26). Although the sample sizes examined are small and results are equivocal, FNMTC does not appear to be caused by a germline BRAF mutation. However, somatic activation of BRAF may be involved in tumor progression in FNMTC (26,38).
Cavaco et al. excluded 17 candidate genes at the 8p23.1–p22 locus but the sample number was very small, and it remains to be established whether the 8p23.1–p22 locus is associated with FNMTC (34).
Discussion
To the best of our knowledge, this review article is the first to comprehensively summarize the available evidence on the genetics of FNMTC. While many studies have apparently shown good evidence for putative susceptibility genes, subsequent analyses have often contradicted these findings. The disparity in results may be explained.
First there is variation in study designs. In particular, the inclusion criteria for FNMTC differ. Some studies use a criterion of two relatives in a family to diagnose FNMTC, whereas others use a minimum of three first-degree relatives to diagnose FNMTC. Unfortunately, some groups do not describe their criteria for diagnosis at all.
In 2006, Charkes suggested that only 31–38% of members in families with two affected members are likely to carry the familial trait, the rest being sporadic. However, in families with three or more members affected with NMTC, the likelihood of possessing the familial trait is 96% (39). Therefore, many of the studies using a criterion of two affected members to diagnose FNMTC may actually have been analyzing sporadic cases of NMTC rather than true FNMTC cases, and this will have diluted the results. For example, two of the potential FNMTC loci, TCO and NMTC1, have also been found to show LOH in sporadic cases of NMTC (29).
The majority of studies do not use a control group in their analysis. In particular, when looking at individual kindreds, diseased and nondiseased individuals in the kindred should be analyzed to reduce the possibility of a false positive by detecting a different familial trait. A control group of sporadic cases should also be analyzed for association with potential FNMTC candidate loci.
The majority of genetic studies include individuals with benign thyroid disease. While it is accepted that benign thyroid disease is a common occurrence in NMTC, it should be considered that it is also a common condition and does not always progress to a carcinoma. Individuals with benign thyroid disease may not have FNMTC and inclusion of these individuals may dilute the results. Very few of the genetic studies conducted, thus far, have taken this into account.
Another hurdle encountered is the sample size. Many of the studies were conducted on single kindreds with unusual forms of NMTC. These kindreds are rare, and subsequent attempts to validate the results have also used small sample sizes yielding low power studies. However, it is prudent to consider why families with FNMTC appear to be quite rare. Many groups have suggested that FNMTC has an autosomal dominant mode of inheritance with incomplete penetrance and variable expression. This is based on observations of male-to-male transmission and multiple horizontal presentations in siblings (4,6,13,16,21,36). Nevertheless, the low incidence of large FNMTC kindreds could be explained by a heterogeneous mode of inheritance mediated through the action of multiple low penetrance alleles.
If FNMTC does have a polygenic mode of inheritance, genetic linkage analysis is unlikely to yield statistically significant results and, as suggested by Bevan et al., one way to increase the power of these studies would be to stratify FNMTC according to phenotypes (28). There is already evidence that some phenotypes of FNMTC are more associated with particular loci. For example, the NMTC1 locus shows more significant linkage to fvPTC than the commoner PTC, and the fPTC/PRN locus shows more significant linkage to PRN. However, given the rarity of particular phenotypes, subcategorization may in itself be problematic.
Conclusion
The limited literature on the genetics of FNMTC is based on small-scale studies. Based on current evidence, FNMTC is likely to represent a polygenic mode of inheritance. The putative susceptibility genes identified appear to account for only a minority of FNMTC. Large-scale studies with strict inclusion criteria, subcategorization of phenotypes, and the use of emerging molecular screening tests are needed to clarify the ambiguity that currently surrounds the genetics of FNMTC at present. The identification of genes for FNMTC could be utilized in the screening, management, and surveillance of NMTC. This could ultimately improve outcomes in FNMTC, which is considered by many to be a more aggressive disease.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.