
Abstract
Background:
Activating germline mutations of the RET gene cause multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma (FMTC), conditions that are inherited in an autosomal dominant manner. In addition, somatic RET mutations have been identified in a variable proportion (about 30–70%) of sporadic (nonfamilial) MTC cases.
Methods:
We describe a Greek family with two novel likely pathogenic sequence variants of the RET gene. The first is a C to T transition at position 2458 (c.2458C>T) that causes an arginine to cysteine substitution (p.R820C) in exon 14 in the intracellular region of the kinase. This sequence variant was identified in an apparently healthy woman who had a recently deceased sister with confirmed aggressive MTC (age of onset 37 years). To assess the pathogenicity of this novel missense sequence variant, screening was performed on all available relatives: her two sons, the mother, and a second sister, including an MTC tumor sample from the deceased sister of the proband. At the time of the investigation, no clinical symptoms suggestive of multiple endocrine neoplasia type 2 or MTC were present in any of the individuals screened.
Results:
The c.2458C>T transition was found in one son, the living sister, and the mother. Interestingly, it was not present in the tumor sample from the deceased sister. Instead, an in-frame deletion of 54 nt in exon 10 resulting in a protein missing 18 amino acids from I590 to G608 (c.1766_1819del 54) was found. Both genetic alterations were present in heterozygous state.
Conclusions:
These data suggest that the novel in-frame deletion was the disease-causing mutation in the deceased sister. The effect of the 2458C>T mutation on the activity of the kinase is under investigation.
Introduction
Materials and Methods
Genomic DNA was extracted from paraffin-embedded tissue using Master Pure Complete DNA and RNA Purification kit (Epicentre, Madison, WI) and from whole blood samples using NucleoSpin Blood (Macherey-Nagel, Düren, Germany) according to manufacturer's instructions. Exons 8, 10, 11, 13, 14, 15, and 16 of the RET gene were polymerase chain reaction (PCR) amplified using Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA). PCR primers and annealing temperatures are shown in Table 1. The PCR products were purified with Nucleofast (Macherey-Nagel) and sequenced with Big Dye Terminator v3.1 according to manufacturer's instructions on a 3730 Genetic Analyzer (Applied Biosystems, Carlsbad, CA). When needed, PCR products were purified from agarose gels with NucleoSpin Extract II (Macherey-Nagel) before proceeding to sequencing. For the genetic identity tests, an ABI AmpFlSTR commercial kit (Applied Biosystems) was used to simultaneously amplify 15 four-nucleotide short-tandem repeat (STR) loci as well as one marker for the XY chromosomes.
Case Report
We report the case of a woman, 36 years old, who was diagnosed in 2006 as having MTC. The patient had a history of a multinodular goiter first diagnosed 1 year prior to the diagnosis of MTC and chronic renal failure due to focal glomerulosclerosis. During follow-up, the dominant thyroid nodule, located in the right lobe, increased in size reaching a maximum diameter of 3.5 cm. Fine-needle aspiration cytology of the nodule was suspicious for thyroid carcinoma. On the basis of these findings, the patient underwent total thyroidectomy and right lymph node dissection. Histology revealed MTC of the right lobe, 3.3 cm in diameter, with lymph node metastases (stage T3N1bM).
At this point the patient was referred to the Endocrine Department for further evaluation and treatment. Serum calcitonin levels were elevated to 1178 pg/mL (normal range below 10 pg/mL) and carcinoembryonic antigen also elevated to 210 (normal range below 5 ng/mL), indicating that residual or metastatic disease was present. Imaging for staging with computed tomography of the neck and thorax showed the presence of residual large necrotic lymph nodes in the right neck (which were considered difficult to surgically dissect), affected lymph nodes in the mediastinum, and bilateral lung metastases. A bone scan revealed bony metastases in the spine. The 131I metaiodobenzylguanidine scintigraphy and 111In-Octreotide scans showed increased uptake in the neck and lungs. No signs or symptoms suggestive of MEN2 syndrome were present in the patient. Twenty-four-hour urine catecholamines, urine vanillyl mandelic acid, and serum calcium were within the normal reference range.
Because of residual local disease, the patient received external neck radiation therapy with 4000 rads. The patient also received long-acting somatostatin analogs for control of diarrhea and flushing. Other treatment modalities, such as therapeutic 131I metaiodobenzylguanidine and chemotherapy, were not provided to the patient because of their known limited therapeutic effectiveness and high possibility of serious side effects in a patient with advanced renal disease. The general condition of the patient deteriorated rapidly and she was in bed most of the time because of weakness. Cancer cachexia appeared and the patient died at 6 months after the diagnosis of the disease.
Measurement of calcitonin and carcinoembryonic antigen was within the normal range in all examined members of the family. Ultrasound of the neck did not reveal any abnormalities in the thyroid gland. No signs or symptoms suggestive of MEN2 syndrome were present in the family. A stimulated calcitonin measurement was considered for all family members, but they refused.
Histology
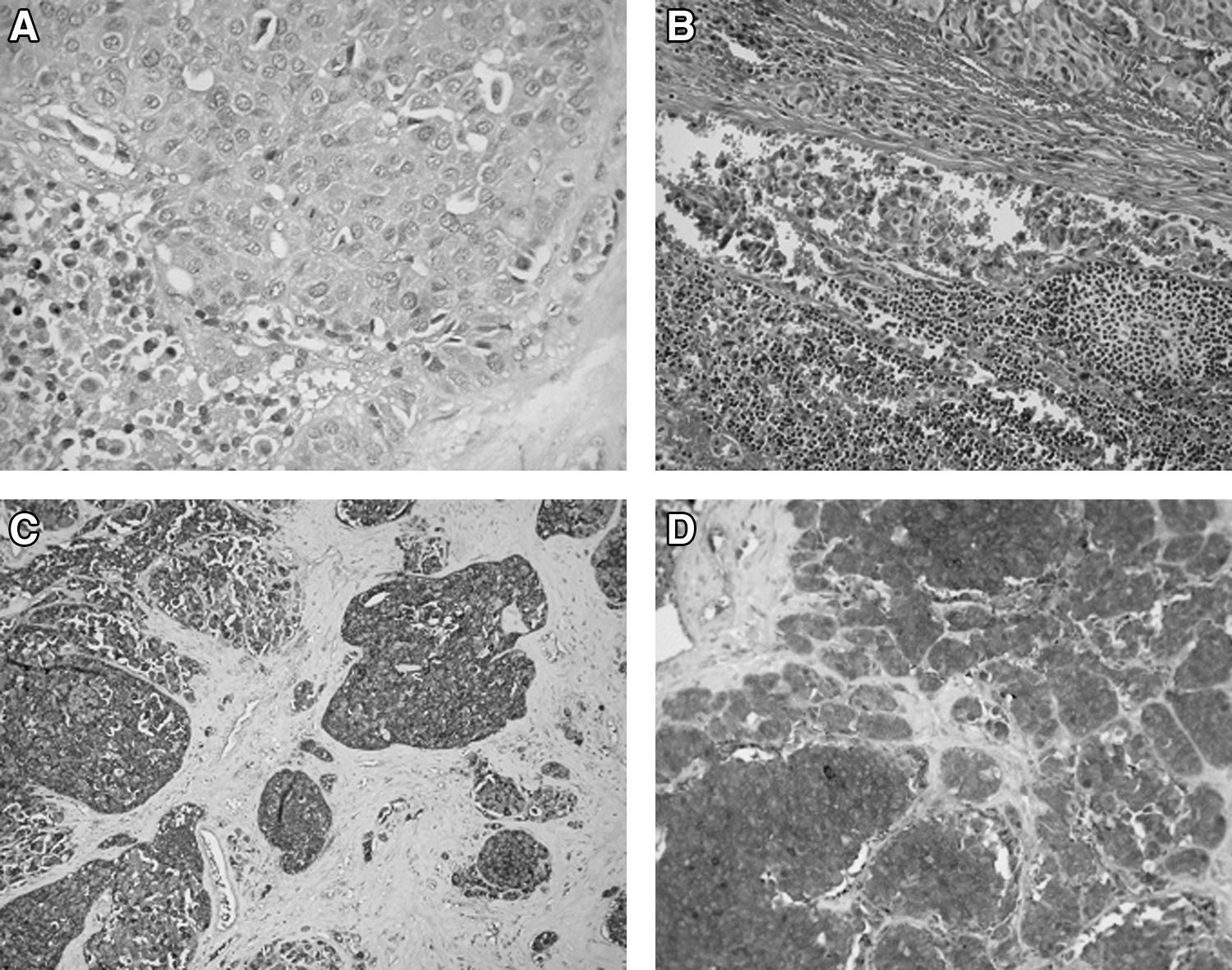
Histology revealed a medullary thyroid tumor of 3.3 cm in maximum diameter, consisting of oval irregular cells (Fig. 1A–D). These tumor cells were arranged in small and large clusters and showed moderate atypia. Lymph and blood vessels of the tumor were infiltrated by neoplastic cells and the tumor showed a few areas of necrosis. The thyroid capsule was invaded by neoplastic cells, which infiltrated the surrounding fatty tissue and small blood vessels. Fourteen out of the 17 lymph nodes showed metastases from the MTC. Immunohistochemistry of the tumor was positive for chromogranin, neuron specific enolase, synaptophysin, and calcitonin.
(
RET Mutational Analysis
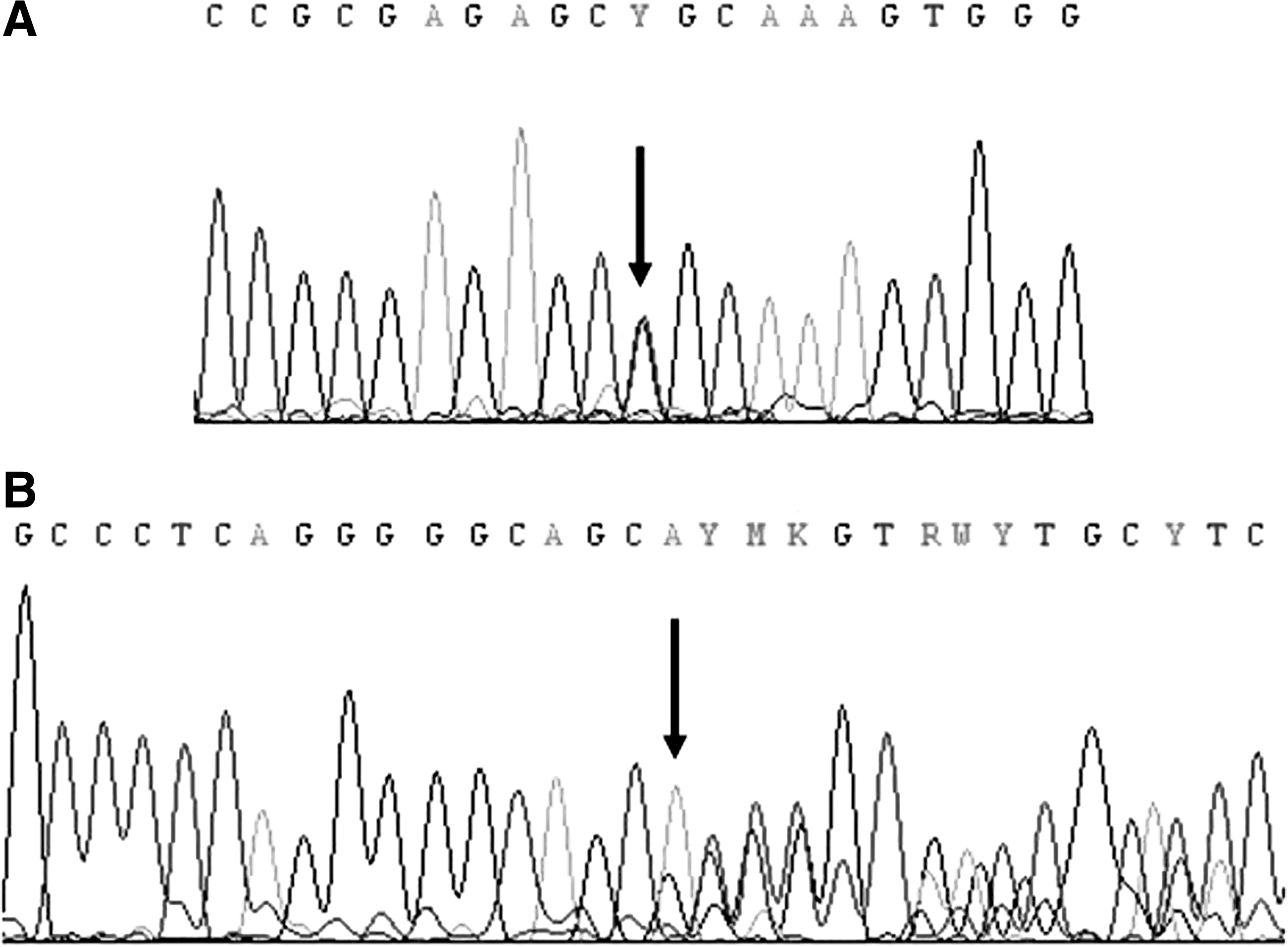
Family member II.2, one of the sisters of the deceased patient (43 years old; Fig. 2), was referred to our laboratory for sequencing analysis of the RET proto-oncogene. Sequencing analysis of exons 8, 10, 11, and 13–16 of the RET gene revealed a novel missense sequence variant, c.2458C>T, which causes an arginine to cysteine substitution (p.R820C) in exon 14 in heterozygous state (Fig. 3A). The same sequence variant was also found in one of her sons (21 years old, member III.1; Fig. 2), her living sister (40 years old, member II.4; Fig. 2), and her mother (72 years old, member I.2; Fig. 2). The potential presence of the novel p.R820C and/or additional alterations was subsequently investigated by PCR amplification of exons 8, 10, 11, and 13–16 using DNA from the paraffin-embedded tumor sample of the deceased sister (member II.3; Fig. 2). Notably, the p.R820C sequence variant in exon 14 was not detected. Instead, PCR amplification of exon 10 revealed the presence of a band shorter than that obtained with germline DNA as shown in Figure 4. The abnormal PCR product, gel purified and sequenced, showed a novel in-frame deletion of 54 nt (c.1766_1819del 54) in heterozygous state, resulting in a protein missing 18 amino acids from I590 to G608 (Fig. 3B). In view of these results, DNA from the patient's (member II.3) paraffin-embedded uninvolved thyroid tissue sections were PCR amplified and sequenced for exons 10 and 14 of the RET gene. The samples were found negative for the presence of the two genetic alterations, strongly indicating that the c.1766_1819del 54 deletion is a somatic mutation.
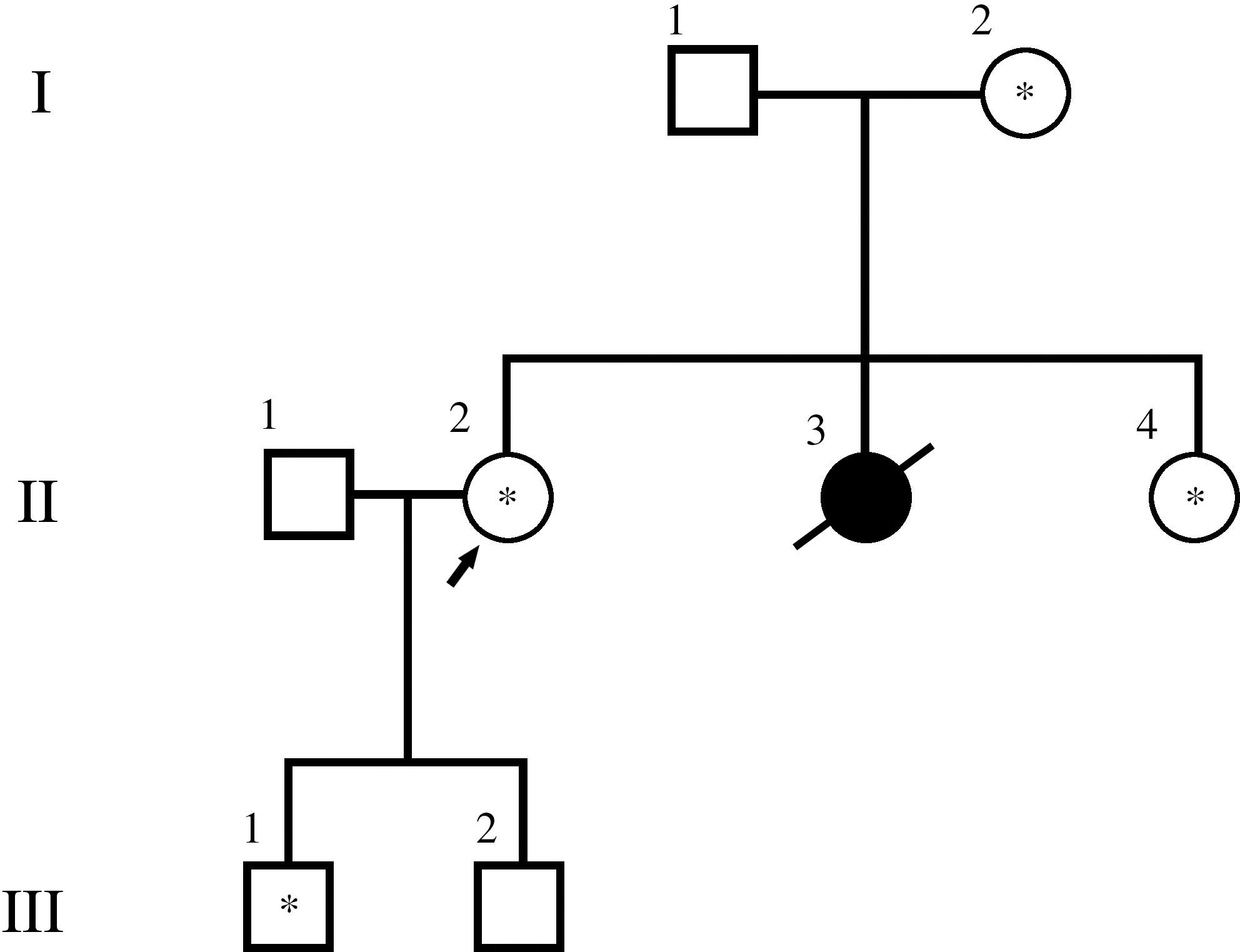
Pedigree of the family showing affected members and members carrying the novel mutation p.R820C (indicated with an asterisk). I, II, and III correspond to three successive generations. I 1 and I 2 are the father and the mother of the three sisters. II 2, 3, and 4 are the three sisters. II 1 is the husband of sister II 2. III 1 and 2 are the sons of II 1 and II 2.
(

Polymerase chain reaction amplification of the region of RET exon 10 in a paraffin-embedded thyroid tumor sample (lane 2), showing a wild-type allele (355 bp) and the allele with a 54 nt in-frame deletion (lane 1, 100 bp DNA ladder).
Further, to establish that the two genetic alterations detected actually occurred within the same family, genetic identity tests were performed using the blood (germline DNA) from the two sisters, tissue (somatic DNA) from the third sister (our patient), and blood (germline DNA) from their mother, but not from the father as he was deceased 4 years ago. The genetic patterns of the four individuals were compared. The results of the genetic analysis are presented in Table 2. Alleles of the three sisters were successfully traced to maternal blood sample at all 15 STR loci. In addition, sister II.3 carrying the deletion shares the paternal alleles at 9/15 loci (60%) with her sisters II.2 and II.4 (Table 2) both carrying the p.R820C sequence variant.
Analysis was performed on DNA extracted from blood of family members II.2, II.4, and I.2 and from paraffin-embedded tissue of individual II.3. Father's DNA was unavailable. Last column shows assumed paternal alleles as derived from the alleles traced in the mother and her three daughters.
STR, short-tandem repeat.
Discussion
Herein we report the identification of two novel genetic alterations in the RET gene within the same family, the c.2458C>T (p.R820C) germline mutation in exon 14 in unaffected members and the c.1766_1819del 54 (p.Iso590_Gly608del) somatic in-frame deletion in exon 10 in an MTC-affected member. Specifically, the mother and her two daughters, individuals I.2, II.2, and II.4 (Fig. 2), respectively, were positive for the germline missense p.R820C sequence change, whereas the third daughter (member II.3; Fig. 1), affected and deceased, was negative for this change and positive for the somatic p.Iso590_Gly608del mutation. This latter mutation was detected in the patient's paraffin-embedded tumor tissue but it was not detected in the patient's uninvolved thyroid tissue. Both genetic changes were in heterozygous state. Further, to establish that the two sequence changes originated from the same family, we performed genetic identity analyses including the mother and her three daughters by using the AmpFlSTR kit comprising 15 markers. The father died 4 years ago; therefore, he was not included in the analysis. Alleles of the three sisters were successfully traced to maternal germline DNA at all 15 STR loci. The deceased affected sister (member II.3) bearing the p.Iso590_Gly608del deletion shared 60% (9/15) of the alleged paternal markers with her sisters II.2 and II.4 bearing the germline p.R820C sequence variant. According to literature, the sharing of paternal DNA may vary between 30% and 69% (8) among sibpairs; therefore, the probability of the three sisters in this family sharing the same biological father cannot be excluded. This inheritance pattern of the variant alleles strongly suggests that the two genetic alterations identified occur within the same family.
The p.Iso590_Gly608del somatic in-frame deletion could possibly explain the severity of the phenotype of the affected member of the family. The patient died at 6 months after the diagnosis of the disease. Because of chronic renal failure, established treatment modalities were contraindicated in that patient. Tyrosine kinase inhibitors targeting activating RET, now used in clinical trials (9), were not available at that time in our hospital. A similar deletion of 48 nt in exon 10 comprising codons 592 to 607 of the RET protein has been previously described in an MTC tumor sample from a 28-year-old woman, who had no family history of thyroid cancer and who presented a 3 cm MTC in the right thyroid lobe and bilateral cervical lymph node metastases (10). This patient was submitted to additional dissection of cervical lymph nodes four times during the next 6 years, because of residual local disease, and she was alive for 9 years after the first diagnosis with elevated calcitonin levels and evidence of distant metastases. An additional somatic in-frame deletion of 27 nt in exon 10 comprising codons 612 to 620 and associated with MTC has also been reported as well as other somatic in-frame deletions in exons 11 and 15 as summarized in Table 3 (10 –13).
The missense c.2458C>T (p.R820C) germline sequence variant in exon 14 residing in the intracellular tyrosine kinase domain of the RET proto-oncogene is a novel finding, and at present its clinical significance is unknown. This is the first time that a genetic change involving codon 820 of the RET proto-oncogene is reported. Arginine at codon 820 of the RET gene is conserved among a diverse range of species including rat, mouse, dog, cow, chiken, mosquito, zebrafish, and Drosophila melanogaster as documented by basic logic alignment search tool analysis involving residue 820 of the RET protein (data not shown). In addition, it has never been detected in the 220 individuals of Greek origin (440 independent alleles) tested for RET mutations and polymorphisms by our group during the last 4 years. These two lines of evidence suggest that the p.R820C sequence change may not be simply a benign polymorphism. On the other hand, family members who carry this sequence variant appear to be healthy at present time with normal calcitonin and negative ultrasound scans. It is, therefore, possible that p.R820C confers a late-onset and a low-grade biological behavior. Based on the above data, the p.R820C is a rare sequence variant of the RET proto-oncogene, and functional studies as well as follow-up of the members of this family will definitely elucidate its biological and clinical significance.
Different RET mutations are related to early development of MTC (6), aggressiveness of MTC (as in our index case), and the presence or absence of other endocrine neoplasms (14). According to the biological behavior of MTC, stratified by mutations of the RET proto-oncogene, the timing and extent of prophylactic surgery can be proposed (15). These genotype–phenotype associations have to be established for rare mutations to define the codon-specific treatment (1,16). Nowadays, DNA testing makes possible the early detection of asymptomatic gene carriers. As the early recognition of the mutant gene carriers gives the possibility of the prevention and cure of MTC, by performing a prophylactic thyroidectomy before the clinical appearance of the tumor, we follow closely all the relatives. Further functional studies are needed to assess the transforming capacity of this novel germline sequence variant. Moreover, we highlight the importance of performing a complete genetic analysis in patients who present with MTC and the available family members.
Footnotes
Disclosure statement
The authors declare that no competing financial interests exist.