
Abstract
Background:
We describe a rare case of congenital hypothyroidism and an extremely high serum thyrotropin (TSH) level caused by a combination of resistance to thyroid hormone (RTH) and a lingual thyroid. As the RTH mutant, R316C, was new, the optimum dose of levothyroxine was unclear. To aid in assessment of the therapy, we characterized the mutant R316C thyroid hormone receptor (TR) and compared it with a common mutant, R316H, using in vitro studies.
Summary:
The patient was a newborn female having severe hypothyroidism with a free thyroxine level of 0.36 ng/dL and a serum TSH level of 177 μU/mL. A scintiscan showed ectopic lingual thyroid tissue without a normal thyroid gland. Supplementation with levothyroxine at a dose of >350 μg/day did not normalize the serum TSH level; however, the patient showed normal growth and intelligence at 14 years of age. Consistent with the results of a computer analysis, the binding of R316C to triiodothyronine (T3) was significantly decreased to 38% that of the wild type. Electrophoretic mobility shift assay demonstrated that like R316H, R316C did not form a homodimer, but formed a heterodimer with RXR. However, a glutathione-S-transferase pull-down assay showed reduced binding of R316C with NCoR in the absence of T3 and impaired release in the presence of T3. In addition, transient transfection experiments demonstrated that unlike R316H, R316C had severe impairment of transcriptional activity on genes both positively and negatively regulated by thyroid hormone. It also had a clear dominant negative effect on genes negatively, but not positively, regulated by thyroid hormone, including the TSH-releasing hormone and TSHβ genes.
Conclusion:
This is the first reported case of a R316C TR mutation. The characteristics of the R316C mutant differed from those of the R316H mutant. Our findings suggest that R316C causes reduced association with and impaired release of NCoR, resulting in RTH predominantly at the pituitary level, and that slightly elevated serum TSH level with high dose of levothyroxine might be optimum for normal growth.
Introduction
Resistance to thyroid hormone (RTH) is a syndrome caused by reduced sensitivity to thyroid hormone (5 –7). More that 2000 affected individuals belonging to about 500 families have been identified, 85% of whom harbor mutations in the thyroid hormone receptor (TR) β gene (THRB) (5). To clarify a mutation of the TRβ gene, the number of the mutated amino acid from the translation start cite flanked by the normal and mutated amino acids is determined. RTH is characterized by an elevated serum thyroid hormone level that is associated with an inappropriately high serum TSH. Therefore, some patients are misdiagnosed as having hyperthyroidism or hypothyroidism, if only the serum thyroid hormone levels or the serum TSH levels, respectively, are considered alone. In most patients, reduced sensitivity to thyroid hormone in peripheral organs is at least partially compensated for by elevated serum thyroid hormone concentrations caused by elevated serum TSH concentrations.
When primary hypothyroidism coexists with RTH, the evaluation of thyroid status is extremely difficult as is the optimum dose of thyroid hormone and optimum TSH level that should be achieved. In fact, such patients are extremely rare. Here we describe the first patient with the RTH mutant R316C. This patient was even more unusual because she was born with a lingual thyroid with severely reduced circulating thyroid hormones at birth. We also characterized the thyroid hormone binding characteristics and functional attributes of the R316C mutant using conventional molecular methods including a ligand-binding assay, a transient transfection analysis, the glutathione-S-transferase (GST) pull-down assay, and electrophoretic mobility shift assay (EMSA) and compared these characteristics with a common TR mutant, R316H.
Patients and Methods
Case report
The patient was a Japanese girl born at 39 weeks to healthy parents. Newborn screening at day 11 after birth revealed a high serum TSH level (>100 μU/mL). She had symptoms of hypothyroidism, including a low growth rate, abdominal distention, mottled skin, open posterior fontanelle, prolonged jaundice (total bilirubin, 16.4 mg/dL), and lethargy. On laboratory analysis, her serum TSH level was 177 μU/mL and free thyroxine (FT4) level was 0.36 ng/dL (normal range: TSH, 0.5–5.5 μU/mL; FT4, 0.78–2.11 ng/dL). Both parents had normal thyroid function. A TSH-releasing hormone (TRH) stimulation test showed an exaggerated serum TSH response (1979 μU/mL at 30 minutes). Treatment with levothyroxine lowered the serum TSH level; however, it could not be maintained below 10 μU/mL.
At the age of 1 year and 4 months without levothyroxine treatment, her serum TSH level was 345 μU/mL and FT4 level was 0.16 ng/dL, indicating severe hypothyroidism. A scintiscan showed ectopic lingual thyroid tissue without a normal thyroid gland (Fig. 1A). We administered triiodothyronine (T3) and a double dose of levothyroxine, but her serum TSH level remained above normal. The detailed doses of levothyroxine and T3 and levels of serum FT4 and TSH are shown in Figure 2. At 11 years of age, a dosage of 300–350 μg/day of levothyroxine was administered. This maintained her serum TSH level below 5 μU/mL. Her serum TSH level was 2.51 μU/mL, FT4 level was 3.2 ng/dL, total cholesterol level was 156 mg/mL, and creatine phosphokinase (CPK) level was 166 IU/L. Her bone age was 10.6 years, and her Tanner breast stage was 2–3. Her IQ was 103. Her growth curve was normal (Fig. 1B), and she showed no signs of hypothyroidism.
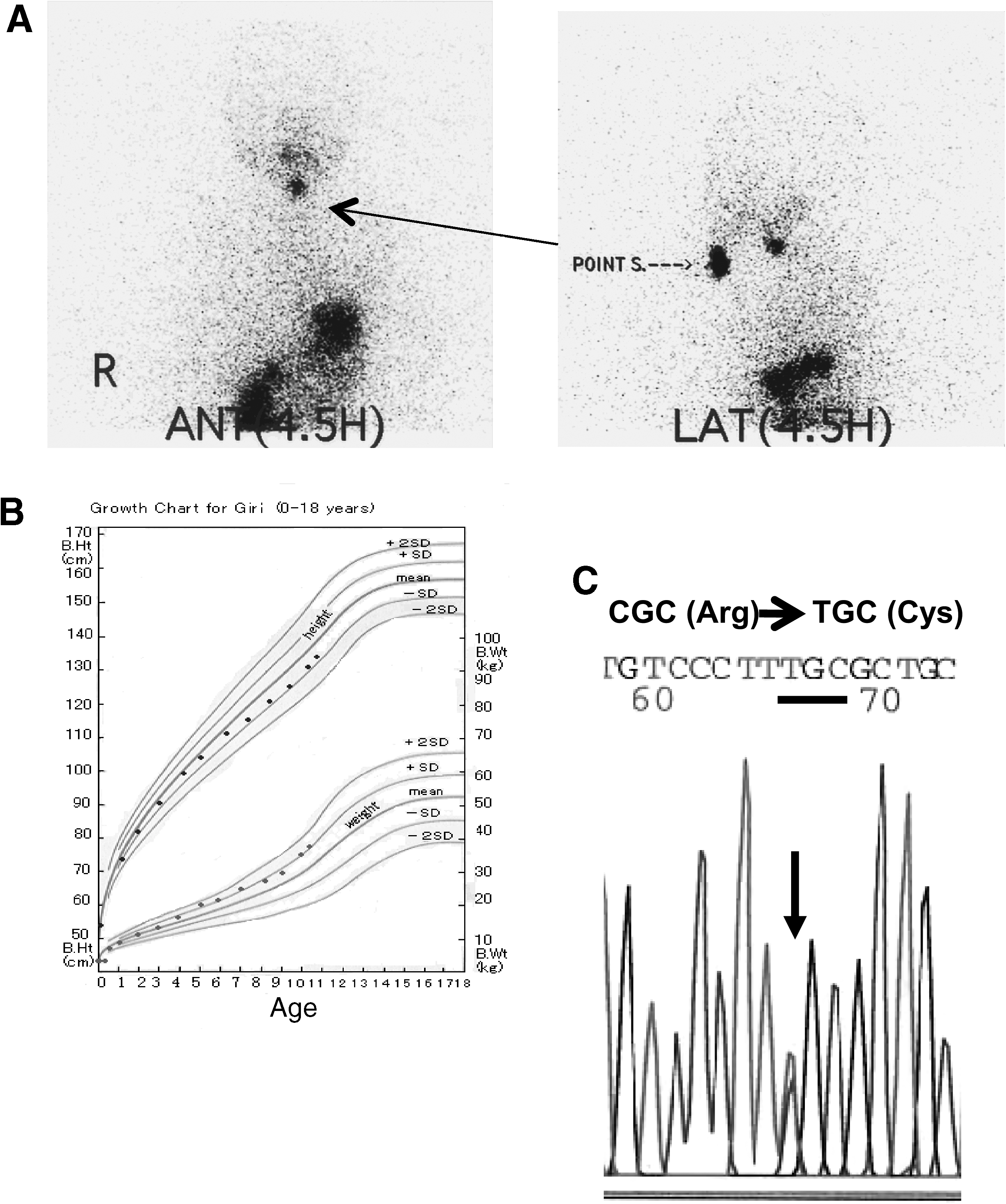
Profile of a patient with resistance to thyroid hormone and a lingual thyroid. (

Clinical course of doses of levothyroxine and triiodothyronine and serum levels of free T4 and TSH. Treatment with levothyroxine and triiodothyronine lowered the serum TSH level; however, it was very difficult to maintain it below 10 μU/mL. At 11 years of age, a dosage of 350 μg/day of levothyroxine was administered. This maintained her serum TSH level below 5 μU/mL. The shadow indicates normal range. TSH, thyrotropin; T4, thyroxine.
Genetic studies
With informed consent, genomic DNA was extracted from leukocytes of the patient and her parents. The study protocol was approved by the ethics committees of the Gunma University School of Medicine. The entire coding exons of the human TRβ gene were sequenced using an autosequencer (PRISMTM 310; Applied Biosystems).
Mammalian cell culture and transfection
CV-1 cells and HeLa cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, as described previously (8). Twenty-four hours before transfection, cells at subconfluence were split into six-well plates. The transient transfection was performed using a calcium phosphate precipitation method, as described previously (8). The total amount of transfected plasmid was adjusted by adding an empty expression vector in all experiments. Sixteen hours after transfection, the medium was changed to Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum treated with AG1-X8 resin (Bio-Rad) and activated charcoal (Sigma) to remove thyroid hormones. Cells were further incubated in the absence or presence of T3 (Sigma).
Plasmid construction
The mutant human TRβ1 cDNAs (R316C, R316H, and R320H) were prepared by PCR mutagenesis and were subcloned into the vector pKCR2. TRH-luciferase (Luc) contains 790 bp of the 5′ flanking sequence and 54 bp of exon 1 from the human TRH gene in pA3-Luc (TRH-Luc). The human TSHβ-Luc contains 1192 bp of the 5′ flanking sequence and 37 bp of exon 1 (TSHβ-Luc). As the promoter activity did not occur with TSHβ-Luc alone, we co-transfected Pit1 and GATA2 for sufficient TSHβ promoter activity (9). Expression vectors for human Pit1 and mouse GATA2 were prepared by PCR and verified by sequencing the DNA. The amplified cDNA was subcloned into the vector pcDNA™3.1 D/V5-His-TOPO.
Binding experiments with T3
Mutant and wild-type TRs were transcribed and translated using a TNT-coupled reticulolysate system (Promega). Affinity for T3 was determined using a filter-binding assay, as reported previously (8). Computer analysis of the binding of T3 with the mutant TR and R316C mutants was done with Swiss-PdbViewer.
Electrophoretic mobility shift assay
The EMSA was performed using radiolabeled thyroid hormone response element (TRE) DR4 or TRE palindrome fragments (8). The consensus sequences used as TRE DR4 and palindrome were 5′-agcttcaggtcacaggaggtcagagag-3′ and 5′-aagattaaggtcatgacctgaggaga-3′, respectively. Double-stranded oligonucleotides were labeled with [α32P]dCTP by a fill-in reaction using a Klenow fragment of DNA polymerase I. The binding reaction, gel electrophoreses, and autoradiography were performed under conditions described previously (8).
GST pull-down assay
[35S]-methionine-labeled wild-type and mutant TRβ were synthesized by in vitro transcription/translation from pKCR2-TR, R316C, R316H, and R320H using T7 RNA polymerase and the TNT-coupled reticulocyte lysate system (Promega Corporation). The synthesis of proteins of expected molecular weights was confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). A cDNA fragment encoding the receptor interaction domain of NCoR and SRC-1 was amplified by PCR using pKCR2-NCoRI and pKCR2-SRC-1 as a template and subcloned in-frame into pGEX4T1 to yield GST-fused proteins in Escherichia coli DH5α. The GST-fused proteins were purified on glutathione–agarose beads (Sigma) and analyzed by SDS-PAGE. Interaction assays and autoradiography were performed as described previously (10). Bound protein was quantified using a Molecular Imager FX (Bio-Rad).
Luc assay
Determination of Luc activity was performed as described previously (8). First, cell monolayers were rinsed twice with phosphate-buffered saline and then lysed with 300 μL of 25 mM glycylglycine (pH 7.8) containing 15 mM MgSO4, 4 mM EGTA, 1 mM dithiothreitol, and 1% (v/v) Triton X-100. Cells were scraped from the dishes and centrifuged at 12,000 g for 5 minutes at 4°C. Assays for Luc activity were performed using 150 μL aliquots of cell lysate and 210 μL of 25 mM glycylglycine (pH 7.8) containing 15 mM MgSO4, 4 mM EGTA, 3.3 mM KPO4, 1 mM dithiothreitol, and 0.45 mM ATP. The reaction was initiated by addition of 200 μL of 0.2 mM
Small-interfering RNA against NCoR
Pooled siRNA oligonucleotides targeting NCoR were designed and synthesized at Dharmacon Research (siGENOME SMART pool NCOR1, L-003518-00). Pooled unrelated siRNA (siCONTROL Non-Targeting siRNA pool, D-001810-0X) was used as a control. These siRNAs were introduced into HeLa cells by the lipofection method (Lipofectamine RNAiMAX™; Invitrogen). Twenty-four hours after the first transfection, a transient transfection of TRH-Luc was performed using calcium phosphate precipitation. Cells were further incubated in the absence or presence of T3.
RNA extraction and real-time PCR
Total RNA was prepared from HeLa cells using ISOGEN (Nippongene), according to the manufacturer's instructions. The cDNA was than reverse-transcribed from 300 ng of total RNA (TaqMan Reverse Transcription Reagents; Applied Biosystems), and 0.5 μL was subjected to real-time PCR. All reactions were performed in triplicate using TaqMan probes and an Applied Biosystems 7500 sequence detection system. TaqMan probes for NCoR (Hs 0019620) and GAPDH (Hs 99999905) were purchased from Applied Biosystems. To determine the expression level of each mRNA, the relative quantification method was performed as described in ABI User Bulletin No. 2. This experiment was repeated at least twice.
Western blot analysis
HeLa cell lysate was prepared in RIPA buffer (1× phosphate-buffered saline, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS), 100 mg/mL PMSF, 30 mL/mL aprotinin, and 1 mmol/mL sodium orthovanadate and then passed through a 23-gauge needle on ice. Cell extracts were incubated on ice for 30 minutes in RIPA buffer. Insoluble cell debris was removed by centrifugation at 10,000 g for 10 minutes. Aliquots of protein-containing supernatant were stored at −80°C. Protein concentrations were determined by the Bradford method using the Bio-Rad protein assay reagent (Bio-Rad Laboratories, Inc.). The lysate (30 μg) prepared above was resolved by SDS-PAGE (10% gel) and transferred to a polyvinylidene fluoride membrane (Hybond-P; Amersham Biosciences) with a semidry system (BIO CRAFT) for detecting the proteins. The blots were blocked for 1 hour with 5% skim milk in Tris-buffered saline with 0.1% Tween 20 and probed for 16 hours with a primary antibody against NCoR or GAPDH. The antibody against GAPDH (sc-20357) was purchased from Santa Cruz Biotechnology, Inc. The antibody against NCoR was kindly provided by Prof. J. Wong (East China Normal University, China). After three washes with Tris-buffered saline with 0.1% Tween 20, antigen–antibody complexes were detected using a peroxidase-conjugated secondary antibody and an enhanced fluorochemiluminent system (ECL-plus; Amersham Biosciences).
Statistical analysis
Statistical analysis was performed using ANOVA and Student's t-test, or the Wilcoxon/Kruskal–Wallis test, with JMP5.1.2 (SAS Institute, Inc.).
Results
Genetic analyses
Direct sequencing of exon 9 of TRβ in the proband indicated that the patient was heterozygous for a novel single-nucleotide substitution, C to T, at nucleotide 1231. This mutation results in the replacement of arginine with cysteine in codon 316 (R316C) (Fig. 1C). Both parents had normal TRβ sequences, indicating that the mutation in the proband was sporadic.
The R316C TR mutant showed reductions in T3 binding and homodimerization on DNA
It has been reported that a cluster of arginines (Arg282, Arg316, and Arg320) in TRβ electrostatically pair with the ligand (T3)'s carboxylate. As shown in Figure 3A, computer analysis revealed that compared with that of the wild-type receptor, the ligand-binding pocket of the mutant receptor is enlarged by the change from Arg to Cys at residue 316. The substitution also causes a reduction in the receptor's electrostatic potential: the mutant receptor should have less electrostatic potential because the pK a of cysteine (∼10) is less than that of arginine (∼12).

Characterization of the R316C mutant TRβ. (
We confirmed this result using synthetic TR and mutant TRs with TNT systems, and Scatchard plot analysis of the binding of T3 revealed that the affinity of R316C was 38% (1.8 × 10−3 M) that of the wild-type TR (3.1 × 10−3 M), similar to the affinity of R316H (1.8 × 10−3 M). The R320H mutant showed a slight reduction (62%) in affinity for T3 of the wild type (Fig. 3B).
EMSA demonstrated that the ability of the R316C mutant to form a heterodimer with RXR was similar to that of R316H, R320H, and wild-type TR on both PAL (palindromic) (TR/RXR, left panel) and direct repeat 4 (DR + 4) (TR/RXR, right panel). However, as shown in Figure 3C, homodimerization of R316C and R316H was significantly impaired on the PAL TRE (TR/TR, left panel), whereas R320H showed a slight decrease in homodimerization.
Transcriptional activity of the R316C mutant
To characterize the functional properties of the R316C TR, we compared its ability to modulate the expression of positively and negatively regulated reporter genes in CV1 cells with that of the wild-type TR and the TR mutants R316H and R320H. In the first instance, we assayed the ability of the mutant receptors to activate the transcription of PAL-TK-Luc containing a palindoromic TRE. As shown in Figure 4A, we found that although the wild-type TR activated PAL-TK-Luc in a dose-dependent manner, R316C and R316H were significantly impaired in the ability to activate transcription. In contrast, R320H displayed only minor impairment.
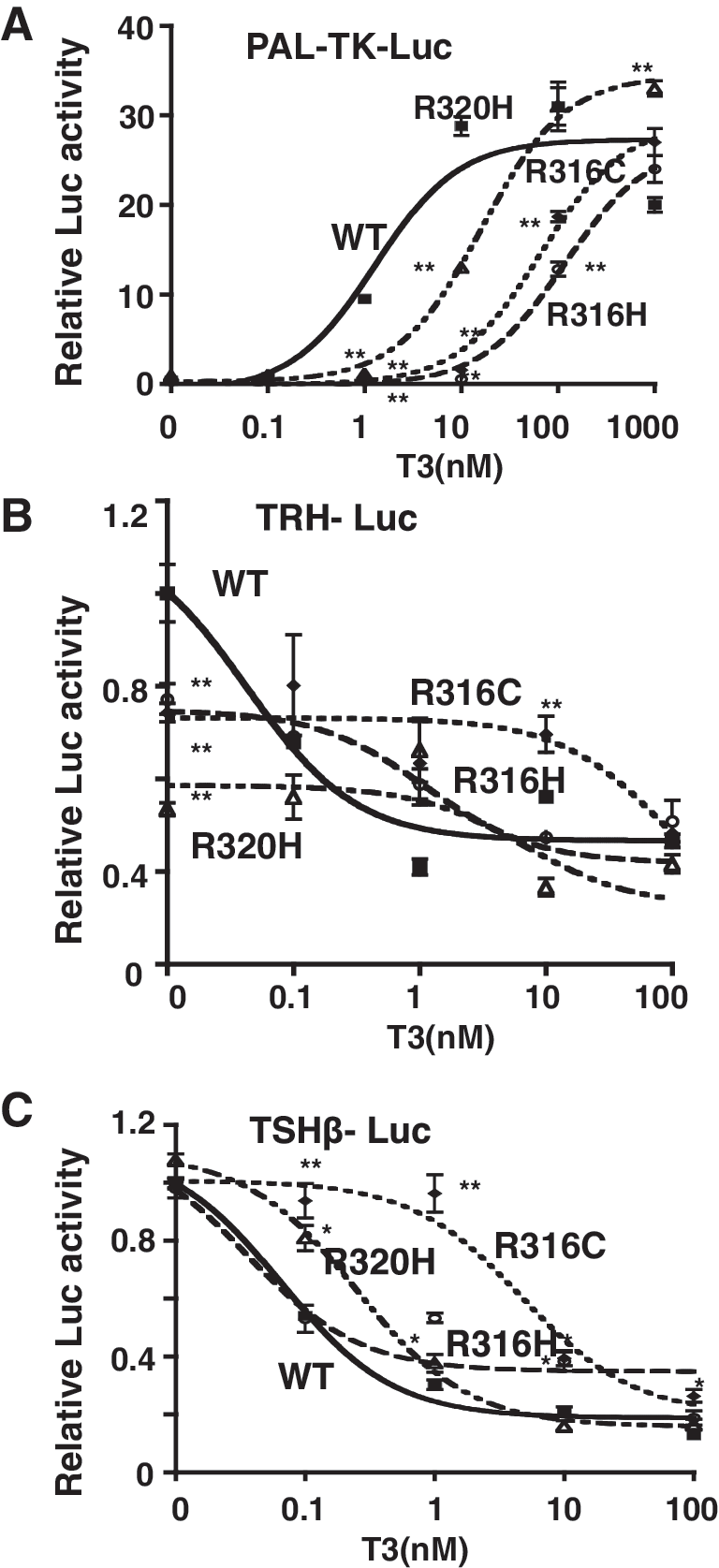
Transcriptional properties of the mutant receptors. The transcriptional properties of wild-type and mutant TRs toward the positively regulated gene (
We next examined the effect of the mutants on the repression of TRH-Luc and TSHβ-Luc expression by thyroid hormone. In TRH-Luc, the activation of the promoter in the absence of T3 by R316C and R316H was ∼80% of the wild-type level and that by R320H was only 50% (Fig. 4B). The addition of T3 inhibited this activity in a dose-dependent manner in the wild-type TR, with ∼90% repression at 100 nM of T3. Although R316H and R320H showed almost normal repression at 0.1–100 nM of T3, the R316C mutant showed severe impairment of the suppression by thyroid hormone, with a level of only 15%–20% that of wild-type TR at 1–10 nM T3 in the TRH gene (Fig. 4B). In the TSHβ promoter, ligand-independent activation by R316C, R316H, and R320H was similar to that by the wild-type TR. As in the TRH promoter, only R316C showed a significant impairment of the repression by thyroid hormone, with almost no repression at 1 nM T3 (Fig. 4C). However, at 10 nM T3, the repression of TSHβ-Luc was ∼80% of that of the wild type. Therefore, the impairment of the inhibition of the TRH and TSHβ genes by T3 in the presence of R316C showed a different profile.
SRC-1 and NCoR-binding properties of mutant receptors
As the impairment of transcription by mutant TRs may be attributable to changes in interaction with cofactors, we performed a GST pull-down assay with nuclear cofactors including SRC-1 and NCoR. The assay showed that although both the wild-type TR and R320H associated normally with SRC-1 in the presence of T3, the R316C and R316H mutants were severely impaired in their ability to associate with SRC-1 (Fig. 5A).

SRC-1 and NCoR binding of the mutant receptors. (
With respect to the binding of NCoR, as shown in Figure 4B, the wild-type TR showed significant binding in the absence of T3, and addition of T3 caused an ∼50% release of NCoR. In contrast, in the absence of T3, the binding of R316C with NCoR was ∼70% of that of the wild-type TR, and addition of T3 resulted in no release of NCoR. Similarly, both R316H and R320H bound with NCoR at a level of ∼70% that of the wild type; however, the addition of T3 resulted in a significant release of NCoR (Fig. 5B).
Dominant negative effect of R316C on positively and negatively regulated promoters
As a dominant negative effect is critical to the phenotype and autosomal dominant inheritance of RTH, we investigated the ability to inhibit wild-type receptor action in a dominant negative manner. In these experiments, equal amounts of wild-type and mutant receptors were coexpressed, and reporter gene activities were assayed at various hormone concentrations. As shown in Figure 6A, none of the mutants examined exerted a dominant effect on genes positively regulated by thyroid hormone (PAL-TK-Luc).

Dominant negative properties of the mutant receptors. Dominant negative effects of mutant TRs on positively regulated genes (
In contrast, R316C exerted a significant dominant negative effect on the inhibition of the TRH and TSHβ promoters by thyroid hormone (Fig. 6B, C). In both constructs, significant ligand-independent stimulation was observed in the absence of T3 (left panels). T3 at 100 nM caused a 50% and 90% reduction in the promoter activity of the TRH and TSHβ genes compared with the vehicle in the presence of wild-type TR. However, the same amount of R316C caused only 25% and 60% inhibition, respectively (Fig. 6B, right panels). Therefore, the dominant negative effect of the R316C mutant was greater on the TRH gene than TSHβ gene. However, R316H and R320H exerted no dominant negative effect on the gene.
Role of NCoR in the regulation of the TRH gene by TR and addition of T3
As impairment of the association with NCoR seemed to be related to the ligand-independent activation of the TRH gene by TRs, we examined the effect of the knocking down of NCoR on the TRH gene activation by TR in the absence or presence of T3. As shown in Figure 7C, significant ligand-independent activation of TR was observed for the expression of TR in HeLa cells, which have been reported to express a significant amount of NCoR, and addition of T3 induced a significant reduction to below the basal level. Transfection of siRNA for NCoR led to a reduction of ∼70% in mRNA and 62% in protein after 48 hours (Fig. 7A, B). In this condition, the ligand-independent activation of the TRH gene by TR was significantly reduced by 75%; however, no significant change was observed for repression of the TRH gene by T3. These findings suggested that NCoR may act as a coactivator for the TRH gene, at least in vitro.

Effect of knockdown of NCoR on the TRH gene. (
Discussion
Here we describe a newly identified mutation, R316C, in a rare case of RTH associated with a lingual thyroid. R320H was the first mutation of TRβ reported to be associated with RTH and a lingual thyroid (11). Although in that case, the serum TSH level at birth was high, 350 μU/mL, there were no symptoms of hypothyroidism, the serum thyroid hormone level was normal, and the patient was euthyroid and did not require replacement therapy with levothyroxine in her first 5 years. That patient might have had a lingual thyroid with the presence of orthotopic thyroid tissue, and it has been reported that abnormalities due to R320H are minor and that some patients with R320H experience no symptoms (12 –15). This may explain why thyroid hormone replacement was not required to maintain normal growth in the first 5 years. Therefore, our patient appears to be the first to have severe congenital hypothyroidism at birth with RTH and a lingual thyroid.
Further, a similar adult case in which a P453T mutant TR was associated with Hashimoto disease and hypothyroidism has been reported, the serum TSH level being 140 μU/mL with 0.34 ng/dL (normal, 0.75–1.75) serum FT4 (16). In this case, replacement therapy with 100 μg/day of levothyroxine improved the symptoms of hypothyridism, suggesting that her hypothyroidism was not severe.
The optimum levels of serum thyroid hormone and TSH for children with RTH are not known, but may be inferred from patients with the same mutation. As R316C is a novel mutation, we first referred to cases of R316H. However, R316H did not cosegregate with the RTH phenotype in family members described and some patients with R316H showed a so-called pituitary RTH phenotype, whereas other showed a generalized RTH phenotype (14,17 –19). We therefore characterized the R316C mutant in vitro to speculate on the phenotype of the individual with R316C. Although R316C and R316H showed similar impaired binding with T3, surprisingly, the characteristics of R316C differed from those of R316H. Unlike R316H, the repression of the TRH and TSH genes by thyroid hormone was severely impaired in R316C. R316H had no dominant negative effect on genes positively or negatively regulated by thyroid hormone, as previously reported (20), whereas R316C exerted a significant dominant effect on genes negatively, but not positively, regulated by thyroid hormone.
The interaction of the mutant receptors with cofactors may explain the functional difference between R316C and R316H. Both mutants were found to be defective in their ability to homodimerize and showed a similar impairment of association with a coactivator, SRC-1. Further, both mutants showed a similar mild decrease in association with NCoR. It has been reported that NCoR prefers to interact with the DNA-bound TR homodimer (21,22). Therefore, the partial defects of the R316C mutant in binding with NCoR may be due to the impaired TR homodimerization. Furthermore, R316H normally released NCoR in the presence of T3, but R316C did not. The significance of the interaction of NCoR to the ligand-independent stimulation of the genes negatively regulated by thyroid hormone remains unclear. Therefore, we conducted an experiment to examine the effect of knocking down NCoR on the repression of the TRH gene by thyroid hormone. Reduced expression of NCoR appeared to abolish the ligand-independent activation of the TRH gene by TR in the absence of T3, but not the repression of the gene by T3. These findings suggested that NCoR might act as a coactivator for the TRH gene.
Further, our experiments in vitro showed the dominant negative effect of the R316C mutant to be greater on the TRH gene than TSH gene. Given these findings, the R316C mutant had not only impaired T3 binding but also partially impaired interactions with SRC and NCoR. Considering that NCoR acts as an activator for genes negatively regulated by thyroid hormone, the combined effect of impaired T3 binding and impaired release of NCoR may have caused an increase in the synthesis of TRH and TSH in the hypothalamus and pituitary, resulting in high serum TSH level in the patient.
In conclusion, we describe the first patient with RTH due to R316C TRβ mutation. The properties of R316C differ from the previously reported R316H mutant. The patient is even more unusual because of concomitant primary hypothyroidism due to thyroid aplasia and lingual thyroid.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.