
Abstract
Background:
Sunitinib is a small molecule that inhibits receptor tyrosine kinases, including the vascular endothelial growth factor receptors, and exhibits antiangiogenic and antitumor activity. This molecule has also been reported to cause hypothyroidism at a high frequency, but the mechanism of this is unknown.
Summary:
A 60-year-old woman was administered sunitinib for the treatment of metastatic renal cell carcinoma. One week later, she displayed overt hypothyroidism with an atrophic thyroid and a marked reduction in vascularity as determined by ultrasonography, despite high levels of thyrotropin. In contrast, during the off-periods in the sunitinib treatment cycles, the volume of her thyroid recovered with an increase in vascularity despite a low level of thyrotropin. These results suggest that thyroid function and volume may depend on the vascularity, which is negatively regulated by sunitinib.
Conclusion:
Our case study provides compelling evidence that sunitinib induces hypothyroidism by reducing blood flow via capillary regression and constriction.
Introduction
Patient
A 60-year-old woman was treated with sunitinib for metastatic renal cell carcinoma in our hospital (2). One week later, she displayed overt hypothyroidism (thyrotropin [TSH]: 20.42 μU/mL [normal range 0.38–4.31]; free triiodothyronine: 1.8 pg/mL [normal range 2.1–3.8]; free thyroxine: 0.98 ng/dL [normal range 0.82–1.63]). Both thyroid peroxidase and thyroglobulin antibodies were negative. During the on-period of sunitinib treatment cycles (arrowhead in Fig. 1), ultrasonography revealed an atrophic thyroid, with the depth of right lobe measured at 9.6 mm and that of the left lobe at 5.4 mm (Fig. 2A). Color Doppler analysis further revealed a marked reduction in parenchymal vascularity (Fig. 3A) and a systolic peak velocity in the superior thyroid artery of 8.4 cm/sec, despite an increased level of TSH at 16.9 μU/mL (inset of Fig. 3A). She was started on a replacement therapy with levothyroxine (Fig. 1).
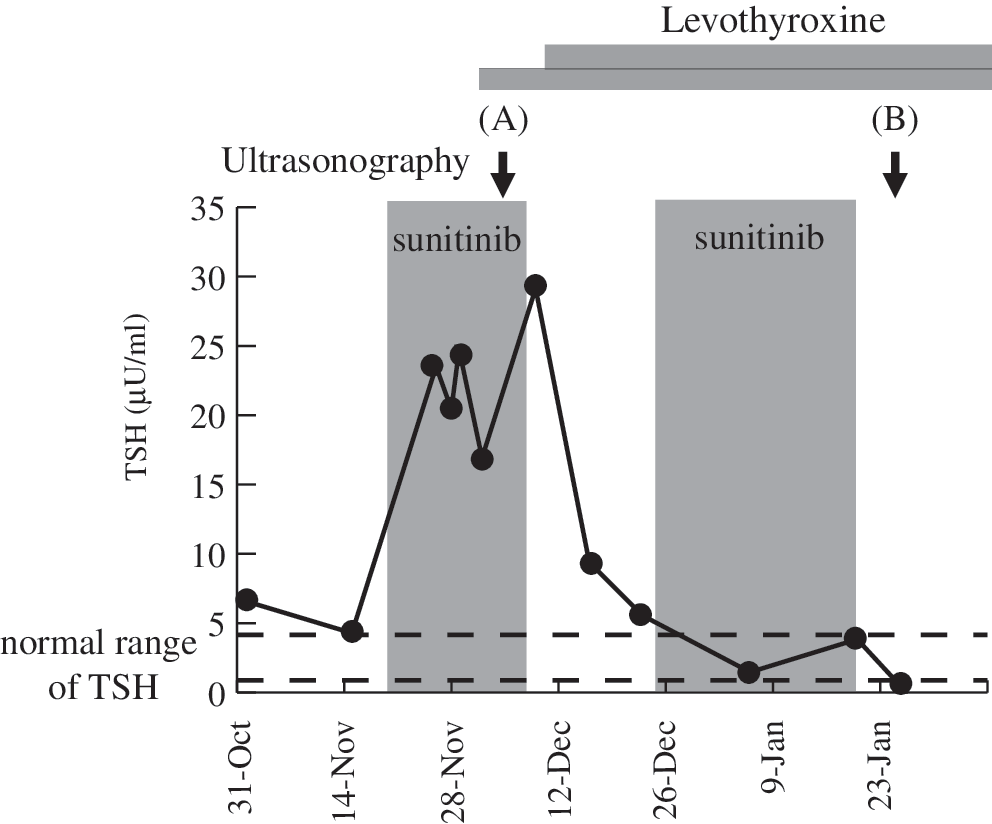
Temporal profile of the thyrotropin (TSH) levels during sunitinib treatment in our patient. Ultrasonography was performed at the arrowed time.
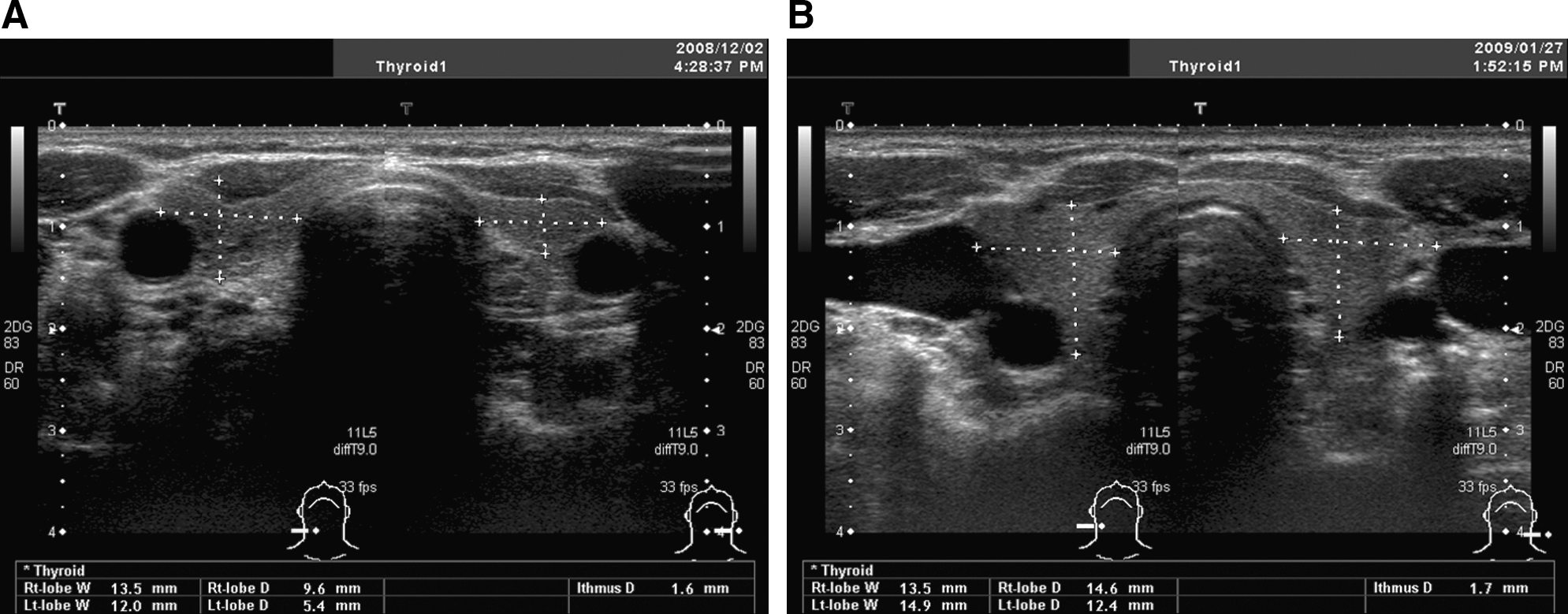
Ultrasonography analysis of the thyroid in our patient at two different time points: sunitinib-on (

Ultrasonography analysis with power Doppler and peak velocity calculations in the superior thyroid artery of our patient at two different time points: sunitinib-on (
Significantly, during the off-period of the sunitinib treatment cycles (arrow in Fig. 1), the volume of her thyroid recovered, with the depth of the right lobe measured at 14.6 mm and that of the left lobe at 12.4 mm (Fig. 2B). There was an increase in the vascularity (Fig. 3B) and also in the peak velocity in the superior thyroid artery (44.2 cm/sec) despite a suppressed level of TSH at 0.26 μU/mL (Fig. 3B, inset). It has been shown that stimulation of the TSH receptor by TSH itself or by TSH receptor antibodies is a determinant of vascularity in the thyroid (3). In our case study, however, the vascularity does not seem to be dependent on the level of TSH but on the period of the sunitinib cycles.
Discussion
Sunitinib-induced thyroid dysfunction can occur in the absence of a predisposition to thyroid autoimmunity (4 –6), which differs from the thyroid dysfunction caused by interferon-α. To date, the pathogenesis of sunitinib-induced thyroid dysfunction has remained unknown, although several mechanisms have been proposed, including destructive thyroiditis (7). However, the frequency of destructive thyroiditis caused by sunitinib is far lower than the incidence of hypothyroidism in patients treated with this drug. In addition, many reported cases, including our current patient, have shown hypothyroidism onset without a thyrotoxicosis phase, suggesting that other causative mechanisms are very possible and should be investigated. In addition, a previous in vitro study has shown that sunitinib may inhibit the synthesis of thyroid hormones by suppressing thyroid peroxidase activity because it inhibited lactoperoxidase activity (8). In contrast, sunitinib does not inhibit the sodium/iodide symporter (9), although this was hypothesized (4).
It has been demonstrated in previous studies that normal thyroid follicular cells express VEGF and VEGF receptor mRNA (10 –12) and that angiogenesis in the thyroid is dependent on the VEGF signaling pathway (13). Our present case, however, may have developed hypothyroidism too rapidly to be explained only by an inhibition of angiogenesis and vessel destruction. Several studies of VEGF-targeted therapies, however, have noted a decrease in tumor perfusion soon after administration of the agent. This can be viewed as vasoconstriction, which is caused by a decrease in the production of vasoactive intermediates such as nitric oxide and prostacyclins, leading to ischemia (14). Taken together, there are sufficient data to hypothesize that sunitinib causes hypothyroidism by inducing the regression of the gland vascular bed with significant capillary alteration and a reduction in density (15). This has been previously reported in a mouse model treated with another tyrosine kinase inhibitor (16). Our ultrasonography analyses strongly support this hypothesis; that is, a reduced vascularity in spite of an increase in the TSH levels during the on-period of sunitinib may be caused by capillary regression and constriction via inhibition of VEGF signaling pathway. In contrast, the increased vascularity that was observed despite a decrease in TSH during the off-period of sunitinib treatment may have been caused by capillary regrowth and vasodilatation via the release of the VEGF block.
We speculate that the mechanism underlying sunitinib-induced destructive thyroiditis may be equivalent to that responsible for sunitinib-induced hypothyroidism (17). The difference between these pathologies may thus depend on the extent of the ischemia. When thyroid blood flow is more severely suppressed by sunitinib, thyroid epithelial cells may undergo apoptosis, which results in destructive thyroiditis. Possibly, also this may depend on the sensitivity of the host. Although our patient showed a marked change in her thyroid volume, another study (4) did not find a change in thyroid volume in the patients studied, which may also reflect the different sensitivity of the hosts. We thus hypothesize that sunitinib-induced thyroid dysfunction, including destructive thyroiditis and hypothyroidism, is caused by ischemia due to capillary regression and constriction.
Footnotes
Acknowledgments
The authors thank Drs. Atsushi Suzuki, Hiromitsu Matsui, Megumi Fujita, and Kozue Izumi for support.
Disclosure Statement
The authors declare that no competing financial interests exist.