
Abstract
Background:
Activation of the Wnt/β-catenin signaling pathway is implicated in thyroid tumorigenesis, and up to 90% of papillary thyroid cancer (PTC) demonstrate aberrant expression of β-catenin. Nonsteroidal antiinflammatory drugs reverse aberrant β-catenin expression and localization in colon cancer. In this study, we tested the hypothesis that the nonsteroidal antiinflammatory drug sulindac would reverse aberrant β-catenin activity in thyroid cancer cells.
Methods:
β-catenin protein levels were determined in thyroidectomy specimens from six consecutive patients and in three different thyroid cancer cells lines (8505-C, KTC-1, and TPC-1) by immunoblotting. Cells of 8505-C and KTC-1 harbor the BRAFV600E mutation, and TPC-1 has the RET/PTC rearrangement. All cell lines were treated with sulindac (100 μM for up to 72 hours). Protein levels of c-myc and cyclin D1 were detected by immunoblotting, and β-catenin localization was determined by immunocytochemistry in the PTC cell lines. PCCL3 rat thyroid cells that conditionally overexpress either BRAFV600E or RET/PTC were also treated with sulindac.
Results:
All PTC specimens and cell lines expressed high levels of β-catenin protein and displayed aberrant nuclear and cytoplasmic localization of β-catenin. Exposure to sulindac for 48 hours reduced β-catenin expression in 8505-C and KTC-1 cells, but not in TPC-1 cells. Further, sulindac treatment reduced c-myc and cyclin D1 levels in 8505-C and KTC-1 cells, but had no effect in TPC-1 cells. Immunocytochemistry demonstrated that sulindac treatment redistributed β-catenin from the nucleus to the membrane in 8505-C and KTC-1 cells. However, sulindac did not affect β-catenin localization in TPC-1 cells. Finally, sulindac was effective in decreasing β-catenin expression and cellular proliferation in BRAFV600E-overexpressing cells, but not in RET/PTC3-overexpressing cells.
Conclusions:
Taken together, our findings demonstrate that sulindac treatment reverses β-catenin activity in 8505-C and KTC-1 cell lines with the BRAFV600E, but not in TPC-1 cells with the RET/PTC mutation. Future studies should investigate the potential for β-catenin-directed therapy for patients with advanced thyroid cancers.
Introduction
There are several genetic alterations that may contribute to the malignant transformation of PTC. Constitutive activation of the RET proto-oncogene via chromosomal rearrangement is found in 5%–40% of PTC cases in adults (2). Similarly, activating mutations of the serine/threonine kinase BRAF, usually caused by a single valine–to–glutamic acid substitution at amino acid 600, are among the most prevalent alterations observed in PTC (3 –5). Mutations of RAS and TRK are also found in PTC exclusive of BRAF or RET activation. Taken together, these genetic alterations are found in approximately 70% of all PTC cases and play significant roles in thyroid cancer differentiation, proliferation, and invasion (4,6,7). Recent studies have suggested that multiple signaling pathways involved in thyroid cancer tumorigenesis may converge downstream to regulate β-catenin activity (8).
β-catenin is a multifunctional protein involved in cell adhesion and gene transcription. Under normal circumstances, β-catenin resides at the plasma membrane where it associates with E-cadherin and α-catenin to regulate the adherens junction and cytoskeleton. Free cytosolic β-catenin is rapidly recruited to a degradation complex where it is targeted for destruction by the ubiquitin-proteasome (9). Activation of the Wnt/β-catenin signaling pathway inhibits this degradation complex, enabling β-catenin to translocate to the nucleus and promote transcription of c-myc, cyclin D1, matrix metalloproteases, and other Wnt target genes (10 –12). Aberrant expression and localization of β-catenin has been implicated in many human cancers, including thyroid tumors. In fact, from 65% to 90% of PTC have abnormal expression of β-catenin (13 –16). Further, cytoplasmic or nuclear, rather than membranous, β-catenin localization is associated with an aggressive tumor phenotype characterized by dedifferentiation of thyroid cancer cells and loss of sensitivity to RAI (17). As such, targeting aberrant β-catenin expression may prove to be a useful approach in patients with aggressive disease.
Treatment with nonsteroidal antiinflammatory drugs (NSAIDs) reverses abnormal β-catenin expression and localization in animal models of colon cancer and in colon cancer cell lines (18 –20). Consistent with this, several randomized trials demonstrated the efficacy of NSAIDs, such as sulindac, aspirin, and celecoxib, in preventing or regressing colorectal adenomas in humans (21 –24). On the basis of these data, we sought to determine the effect of sulindac in thyroid cancer cell lines. We hypothesized that sulindac treatment would reverse aberrant β-catenin expression and normalize localization back to the plasma membrane. We found this hypothesis to be true only in PTC cells that lack the RET/PTC mutation.
Materials and Methods
Materials
Mouse anti-human β-catenin antibody was purchased from BD Biosciences. Rabbit anti-human cyclin D1, mouse anti-human c-myc, rabbit anti-RET, and anti-BRAF antibodies were obtained from Santa Cruz Biotechnology. Mouse anti-actin antibody, pan Ab-5, was purchased from Neomarker. Secondary horseradish-peroxidase-conjugated anti-rabbit and anti-mouse antibodies and fluorescein anti-mouse immunoglobulin G were obtained from Vector Laboratories. Sulindac sulfide was purchased from Sigma. Doxycycline was purchased from Invivogen.
Cell culture
The human PTC cell line TPC-1 was kindly provided by Dr. Orlo H. Clark (University of California at San Francisco, San Francisco, CA). The human PTC cell line 8505-C was obtained from Dr. Sareh Parangi (Massachusetts General Hospital, Boston, MA). Another papillary thyroid cell line, KTC-1, was obtained from Dr. James A. Fagin (Memorial Sloan Kettering Cancer Institute, New York, NY). PTC3-5 and PC-BRAFV600E cells were also kindly provided by Dr. James A. Fagin (Memorial Sloan Kettering Cancer Institute, New York, NY) and were derived from rat thyroid PCCL3 cells to obtain doxycycline-inducible expression of RET/PTC3 or BRAFV600E, respectively (25,26). Cells were cultured as previously described (27 –29). All cells were subcultured at 80% confluency by trypsinization (in a 0.5% [v/v] trypsin solution, supplemented with 0.2% [v/v] ethylenediaminetetraacetic acid).
Western blotting
Cell lysates were separated by 4%–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrophoretically transferred to a polyvinylidene fluoride (PVDF) membrane (Invitrogen). The membranes were blocked with 5% casein in a Tris-buffered saline–Tween 20 solution. The blots were performed as described previously (30). Proteins on each immunoblot were observed with Western blot chemiluminescence reagent (Amersham Biosciences).
Immunofluorescence staining
Cells were grown and treated on glass coverslips at 37°C. Cells were fixed in 4% paraformaldehyde solution for 30 minutes and blocked in 5% of casein solution. Fifty microliters of mouse anti-human β-catenin antibody (4.5 g/mL in 0.5% fetal bovine serum/0.5% Tween 20/phosphate-buffered saline [PBS]) was subsequently applied as the primary antibody solution followed by overnight incubation at 4°C in a humidified chamber. After extensive rinsing with PBS, coverslips were incubated with 50 μL of fluorescein anti-mouse immunoglobulin G secondary antibody (1:20 in 0.5% fetal bovine serum/0.5% Tween 20/PBS) for 3 hours at 37°C. After three washes with PBS, glass coverslips were subsequently washed three times with distilled water, mounted with Fluoromount G (Electron Microscopy Sciences), and examined using a fluorescent microscope.
MTS proliferation assay
The MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) Assay, Celltiter 96 AQ (Promega), was used to assess sulindac effects on cell viability. Trypsinized cells (5000 cells/well) were seeded in a 96-well plate. Twenty-four hours later, a standard culture medium was replaced with dimethyl sulfoxide or various doses of the sulindac-containing medium. After 48 hours of treatment in a humidified CO2 incubator at 37°C, MTS reagent (20 μL) was added to each well. After 2 hours, absorbance at 490 nm (A490) was quantified using a Spectramax M5 plate reader (Molecular Devices). Response was defined as [(mean drug-treated A490 − blank)/(mean untreated control A490− blank)] × 100.
Human thyroid tumor tissue samples
Human PTC tissues and normal adjacent thyroid tissue were obtained from surgical specimens resected from patients treated at the Brigham and Women's Hospital and were immediately frozen in liquid nitrogen. Patients 1–5 had the classical variant of PTC and patient 6 had the follicular variant of PTC. The tumor samples were stored frozen until the time of protein extraction. This study has been approved by the Institutional Review Board of Brigham and Women's Hospital.
Statistical analysis
A Student's t-test was performed to evaluate differences in cellular proliferation in sulindac-treated versus control dimethyl-sulfoxide-treated cells.
Results
β-catenin is highly expressed in human PTCs
Aberrant localization of β-catenin is found in many human cancers, including thyroid cancers. Prior reports show that up to 90% of PTC demonstrate aberrant nuclear and cytoplasmic β-catenin localization by immunohistochemistry (13 –16). However, none of these studies comment on β-catenin protein levels in PTC, which should be upregulated upon Wnt activation. To investigate β-catenin expression in human thyroid samples, Western blot was performed in six consecutive PTC specimens. β-catenin protein was highly expressed in the six PTCs relative to the normal adjacent thyroid tissue (Fig. 1). This finding confirms activation of the Wnt pathway as demonstrated by increased levels of stabilized β-catenin protein and corroborates prior immunohistochemical findings of Wnt signaling in PTC.

β-catenin protein expression in fresh human thyroid tumor samples. Western blot analysis of β-catenin protein expression in human thyroid tumor samples and adjacent normal thyroid tissue was carried out. Actin expression served as a loading control. Final diagnosis of each specimen was confirmed pathologically. N, normal thyroid tissue; T, papillary thyroid cancer (PTC) tissue from different patients.
Sulindac suppresses β-catenin, cyclin D1, and c-myc protein expression in 8505-C and KTC-1 but not TPC-1 cells
We next examined β-catenin protein expression level in PTC cell lines. Cells of 8505-C and KTC-1 harbor the BRAFV600E mutation, and TPC-1 has the RET/PTC rearrangement. We found that β-catenin was highly expressed in these PTC cell lines (Fig. 2). To test our hypothesis that sulindac would reverse aberrant β-catenin expression in PTC, we treated all three cell lines with 100 μM of sulindac for 48 and 72 hours. In 8505-C and KTC-1 cells, sulindac treatment inhibited β-catenin protein expression levels in a time-dependent manner. In addition, protein levels of cyclin D1 and c-myc, two β-catenin downstream regulated target genes, decreased after sulindac treatment in 8505-C and KTC-1 cells. Sulindac treatment (100 μM) showed no suppressive effects on β-catenin, cyclin D1, or c-myc protein expression levels in TPC-1 cells with the RET/PTC mutation (Fig. 2).

Effect of sulindac on β-catenin, cyclin D1, and c-myc expression levels in PTC cells. TPC-1, 8505-C, and KTC-1 cells were treated with control (DMSO) for 72 hours or sulindac (100 μM) for 48 and 72 hours. Total cell lysates were isolated, and β-catenin, cyclin D1, and c-myc protein expression levels were measured by Western blotting. Actin was used as a loading control. DMSO, dimethyl sulfoxide.
Sulindac induces β-catenin relocalization in 8505-C and KTC-1 but not TPC-1 cells
To determine the effect of sulindac on β-catenin cellular localization, we performed immunocytochemistry for β-catenin and E-cadherin in TPC-1, 8505-C, and KTC-1 cells. At baseline, β-catenin was primarily localized in the cell nucleus in all three PTC cell lines (Fig. 3). After sulindac treatment (100 μM) for 48 and 72 hours, β-catenin relocalized from the nucleus to the cell membrane in 8505-C and KTC-1 cells. Staining for E-cadherin demonstrated colocalization at the cell membrane with β-catenin after sulindac treatment as seen on merged images. In TPC-1 cells, however, sulindac treatment (100 μM) had little effect on the localization of β-catenin.

Immunocytochemical analysis of β-catenin localization in PTC cells. TPC-1, 8505-C, and KTC-1 cells were treated with control (DMSO) for 72 hours or sulindac (100 μM) for 48 and 72 hours. β-catenin and E-cadherin staining using appropriate anti-human antibodies followed by fluorescein immunoglobulin G staining was completed. 4′,6-Diamidino-2-phenylindole (DAPI) counterstaining was performed to label cell nuclei. Images were merged to demonstrate β-catenin and E-cadherin colocalization at the cell membrane. Fluorescence microscopy images after each treatment are shown (original magnification, 400 ×). Color images available online at
β-catenin expression is independent of BRAFV600E activity
To determine whether specific genetic mutations were responsible for the variable responses to sulindac in PTC cells, we investigated the role of BRAFV600E and RET/PTC3 overexpression in thyroid cancer cells. We obtained rat thyroid cell lines (PCCL3) that were stably transfected with expression vectors for either BRAFV600E (PC-BRAFV600E) or RET/PTC3 (PTC3-5) and engineered to express the oncoproteins in a doxycycline-dependent manner (25,26). Conditional overexpression of BRAFV600E after treatment with 1 μg/mL of doxycycline significantly enhanced expression levels of BRAF in PC-BRAFV600E cells (Fig. 4A). β-catenin levels were unchanged in the presence or absence of BRAF. Treatment with sulindac (100 μM) for 48 hours suppressed β-catenin expression in PC-BRAFV600E cells in the presence or absence of doxycycline (1 μg/mL). These results demonstrate that BRAF does not play a critical role in the upregulation of β-catenin expression nor does it affect sulindac inhibition of β-catenin levels (Fig. 4A).
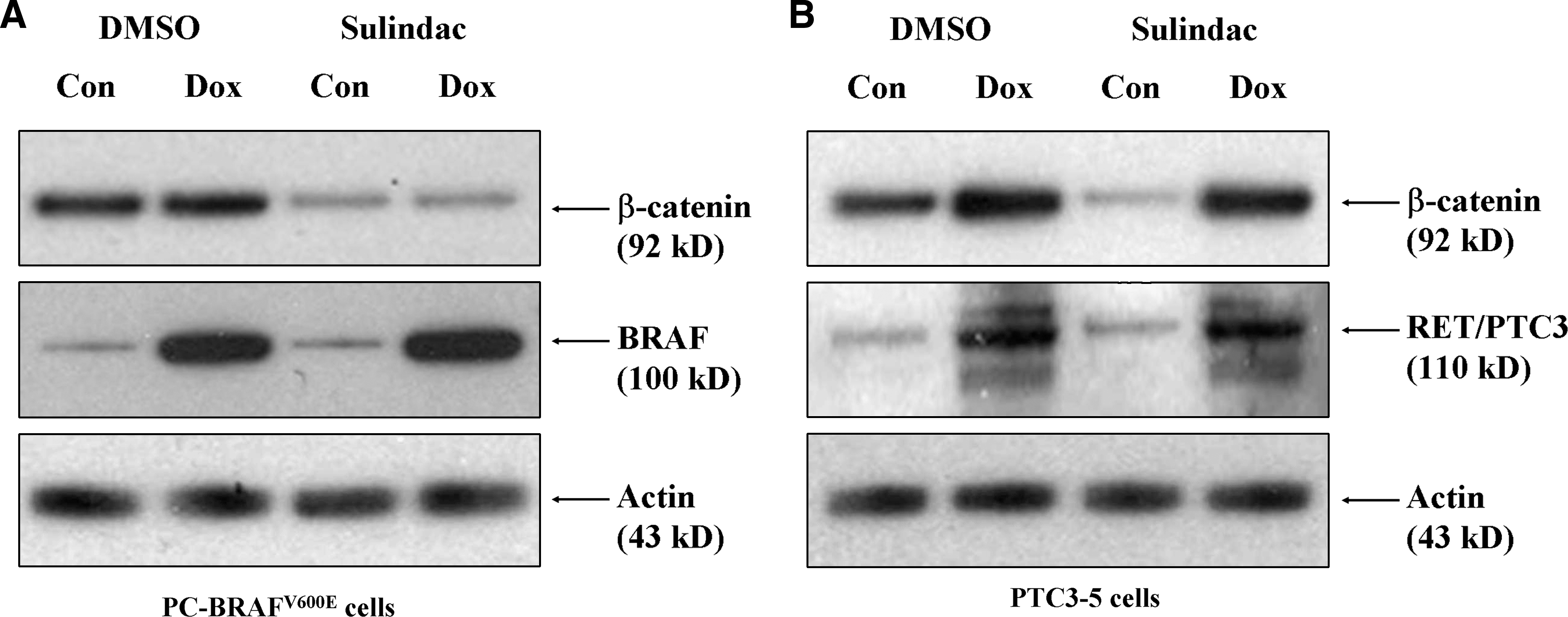
Effect of sulindac on BRAFV600E- and RET/PTC3-overexpressing thyroid cells. PC-BRAF (
RET/PTC overexpression induces aberrant β-catenin expression, which is not reversible by sulindac treatment
We next investigated the specific role of RET/PTC3 rearrangements on β-catenin expression and response to sulindac treatment. Treatment of PTC3-5 cells with 1 μg/mL of doxycycline significantly enhanced both RET/PTC3 and β-catenin expression (Fig. 4B). Sulindac (100 μM) treatment for 48 hours did not suppress β-catenin levels in PTC3-5 cells upon conditional expression of RET/PTC3 with doxycycline (1 μg/mL). This finding suggests that RET/PTC3 upregulates β-catenin expression and confers resistance to sulindac inhibition of β-catenin activity in PTC cells.
Sulindac reverses proliferation in BRAFV600E but not RET/PTC3-overexpressing cells
We next determined if sulindac treatment could reverse cellular proliferation driven by BRAFV600E or RET/PTC3 mutations. Sulindac treatment significantly decreased proliferation in both 8505-C and KTC-1 cells, but not in TPC-1 cells with the RET/PTC mutation (Fig. 5A). Further, conditional overexpression of BRAF in PC-BRAFV600E cells significantly enhanced proliferation, which was reversible by sulindac treatment. Similarly, conditional overexpression of RET/PTC3 in PTC3-5 cells increased proliferation; however, this effect was not reversed by sulindac treatment (Fig. 5B).
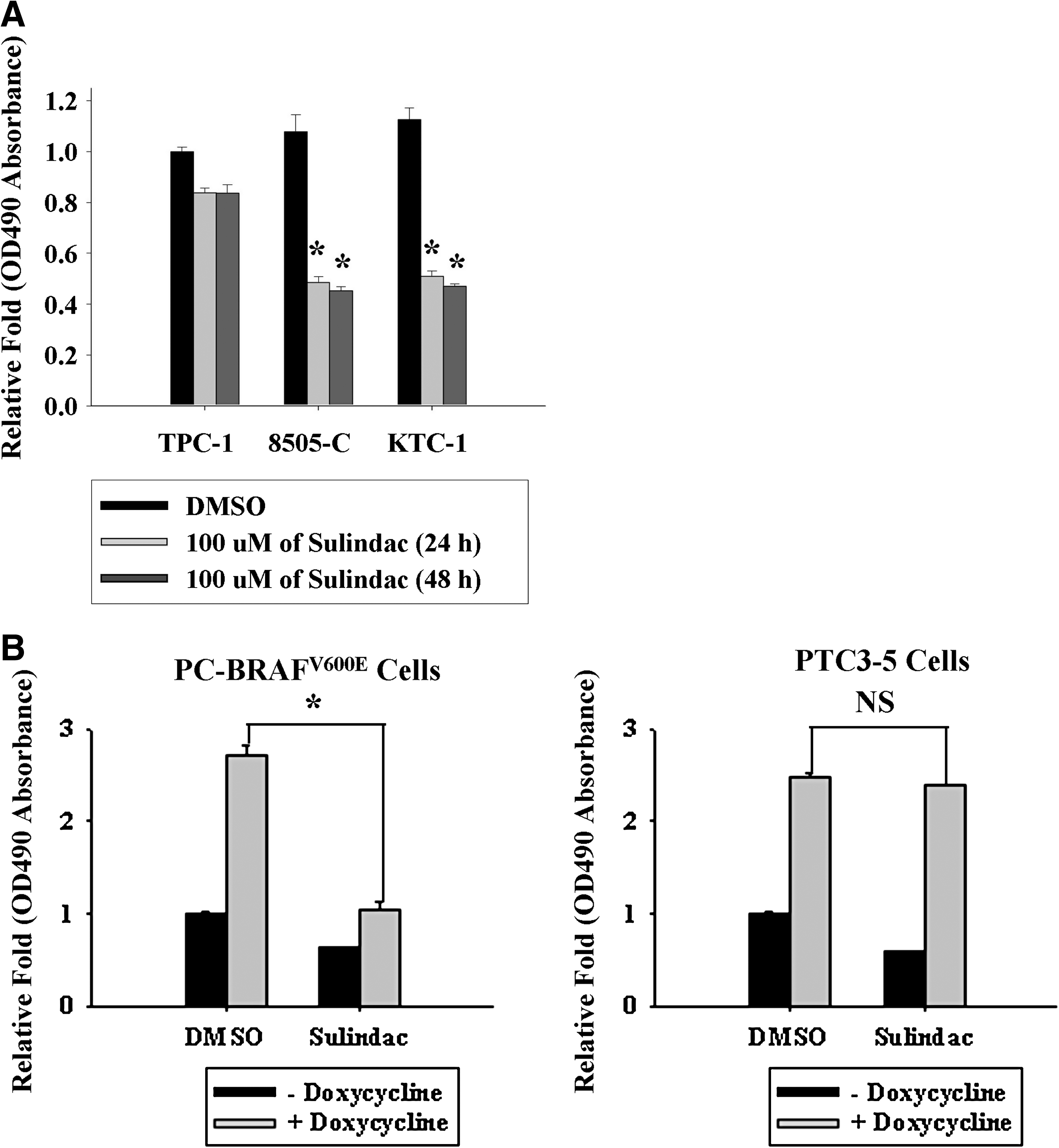
Effect of sulindac on proliferation in PTC, BRAFV600E-overexpressing, and RET/PTC3-overexpressing thyroid cells. Proliferation was assessed in PTC cells (
Discussion
Aberrant β-catenin activity is frequently observed in many cancers, and our findings corroborate prior reports that β-catenin expression and localization are often abnormal in PTC (13 –16). Further, we show that this aberrant activity can be reversed with NSAID therapy in some PTC cell lines. Our data demonstrate that sulindac is highly effective in reversing aberrant β-catenin levels, nuclear localization, cellular proliferation, and expression of the downstream products c-myc and cyclin D1 in 8505-C and KTC-1 cells with the BRAFV600E mutation, but not in TPC-1 cells with the RET/PTC mutation.
Constitutive activation of the RET proto-oncogene via chromosomal rearrangement is a common genetic event found in up to 40% of PTC cases (2). Our results show that RET/PTC positively regulates β-catenin expression level in thyroid cells, consistent with previous reports by Gujral et al. and Castellone et al. (31,32). Gujral et al. recently showed that activated RET kinase may promote tumorigenesis in medullary thyroid cancer (MTC) through direct tyrosine phosphorylation of β-catenin at the plasma membrane (31). This event releases β-catenin from the cytoskeleton, where it can translocate to the nucleus and drive gene expression. Castellone et al. also described the functional interaction between RET and β-catenin by demonstrating that RET activates the PI3K/AKT and Ras/MEK/ERK pathways to stabilize cytoplasmic β-catenin (32). Further, Cassinelli et al. corroborated this crosstalk between RET and β-catenin, and showed that β-catenin nuclear localization could be reversed using the small molecule inhibitor RPI-1 to silence RET/PTC activity (33). Taken together, these data suggest that RET/PTC drives multiple pathways that converge upon β-catenin to promote tumor proliferation and invasion.
Our results demonstrate that sulindac is not effective in reversing nuclear β-catenin localization or protein expression in TPC-1 cells with the RET/PTC mutation. The mechanism for this resistance may be the result of constitutive β-catenin phosphorylation at the cell membrane by RET kinase, allowing β-catenin to translocate to the nucleus and escape cytoplasmic degradation. Thus, RET/PTC cells may bypass any prostaglandin inhibitory effects of sulindac that target β-catenin for proteasomal degradation. In support of this theory, we showed that specific overexpression of RET/PTC3 in PCCL3 cells confers resistance to sulindac inhibition of β-catenin activity (Figs. 4 and 5). Further, sulindac had no effect on inhibiting β-catenin levels in the medullary thyroid cell line MTC 1.1, which has an activating RET mutation (data not shown). Given these findings, patients with RET/PTC mutations may be poor candidates for β-catenin-targeted sulindac therapy, although there may still be potential for NSAID use in this patient population given its prostaglandin-independent antitumoral effects. In fact, Zatelli et al. reported that cyclooxygenase-2 inhibitors can reverse the chemoresistant phenotype of MTC cells in a prostaglandin E2 (PGE2)-independent manner (34). Future studies should investigate prostaglandin-dependent and prostaglandin-independent mechanisms for NSAID inhibition of β-catenin activity, and examine the role of tyrosine kinase inhibitors, such as imatinib, in downregulating β-catenin activity for these specific RET/PTC cancers.
Contrary to our findings with RET/PTC cells, sulindac treatment had a profound effect on reversing β-catenin localization, protein levels, and proliferation in 8505-C and KTC-1 cells with the BRAFV600E mutation. To our knowledge, our study is the first to show that NSAIDs can inhibit aberrant β-catenin activity and localization in PTC cells. BRAFV600E is known to induce thyroid tumorigenesis in cells and transgenic mice through activation of the RAS/BRAF/MEK/ERK pathway (35). Several studies have also suggested that tumors with the BRAFV600E mutation have an aggressive phenotype characterized by increased tumor invasion, perhaps via expression of matrix metalloproteinases, decreased expression of thyroid-specific genes, and increased risk for recurrence (36 –39). As such, reversal of β-catenin activity by NSAIDs may reduce the invasive or metastatic potential of BRAFV600E tumors by inhibiting expression of cyclin D1, matrix metalloproteinases, c-myc, and other oncogenic factors. Further, Castellone et al. showed a direct link between PGE2 expression and increased β-catenin activity in colon cancer cells, suggesting that prostaglandin inhibition may be an important target for drug therapy (40). Further investigation is warranted to determine the effect of NSAIDs on tumor redifferentiation and behavior in patients with advanced disease that is refractory to conventional treatment.
There is a growing body of literature to support the interplay between inflammation and cancer, and NSAIDs such as sulindac may provide antitumorigenic effects through downregulation of cyclooxygenase-2, PGE2, and β-catenin activity as well as through prostaglandin-independent paths. Consequently, NSAID therapy may be a useful adjunctive treatment in patients with aggressive papillary or other types of thyroid cancer, and may open a window of opportunity for further treatment with RAI, small-molecule-directed therapy, or tyrosine kinase inhibitors. A recent phase II clinical trial by Mrozek et al. demonstrated a modest response to celecoxib treatment in 32 patients with metastatic differentiated thyroid carcinoma (41). However, response to treatment was not correlated with histological subtype, and patients were not stratified by genotype. The single partial responder was a patient with PTC, and 12 patients, of unknown histology, were categorized under stable disease. Given the findings from our study, it would be interesting to associate clinical response to celecoxib with histological subtype and molecular characteristics.
Conclusions
Sulindac treatment reverses overexpression and aberrant localization of β-catenin in 8505-C and KTC-1 cell lines with the BRAFV600E mutation, but not in TPC-1 cells with the RET/PTC mutation. Our results highlight the importance of genotyping individual cancers, as some treatment modalities may only be effective in patient populations with specific genetic alterations. Future studies should investigate the potential for β-catenin-directed therapy targeting patients with advanced thyroid cancers and the susceptible genetic makeup.
Footnotes
Acknowledgment
The authors would like to thank Dr. James Fagin for generously providing us with KTC-1, PTC3-5, and PC-BRAFV600E cells.
Disclosure Statement
The authors have nothing to disclose. The authors declare that no competing financial interests exist.