
Abstract
Background:
Congenital central hypothyroidism (CCH) is a rare condition that is often diagnosed in late childhood in countries where neonatal screening programs rely solely on detecting thyrotropin (TSH) elevation. TSHβ gene mutation is one of the causes of CCH. We describe two cases of c.Q49X mutation and three cases of c.C105Vfs114X mutation in exon 3 of the TSH β-subunit gene.
Summary:
We found two different TSHβ gene mutations in two families. In one family, we identified a missense mutation in exon 3 leading to a premature stop at position 49 (c.Q49X) in the two affected twins. In the other family, the three affected siblings had a 313delT nucleotide deletion leading to a frame shift responsible for premature termination at codon 114 (c.C105Vfs114X); neonatal screening showed very low TSH levels in all three patients. The presence of inappropriately low TSH levels at birth in the three affected members of the second family raises questions about the value of the TSH level for CCH screening.
Conclusions:
The marked phenotypic variability in patients with the c.Q49X mutation suggests modulation by interacting genes and/or differences in the genetic background. TSHβ gene mutations should be suspected in neonates with inappropriately low TSH levels.
Introduction
In patients with CCH, co-existing pituitary or hypothalamic disease may lead to deficiencies in other pituitary hormones (1,2,6). Causes of isolated CCH include inadequate hypothalamic-pituitary control or low TSH reserve (e.g., Klinefelter syndrome and depression) (7,8), autosomal recessive thyrotropin-releasing hormone (TRH) deficiency (9), autosomal recessive TRH receptor-inactivating mutations (10), and autosomal recessive TSH deficiency (11).
TSH is a pituitary hormone composed of two linked α and β subunits. The β-subunit differs across glycoprotein hormones (follicle-stimulating hormone, luteinizing hormone, and chorionic gonadotropin) and ensures the specific biological activity of each (12). Isolated TSH deficiency can result from mutations in TSHβ (OMIM No. 188540) or in TRH receptor genes (OMIM No. 188545). Recessive homozygous or compound heterozygous TSHβ gene mutations have been identified in more than 30 families (11). Serum TSH shows both quantitative and qualitative changes during ontogenesis, and the process that results in bioactive TSH remains incompletely understood (13,14). Mutations in the TSH β-subunit may thus result in an immunoreactive TSH molecule that lacks bioactivity (15).
We describe the features of CCH in two sibships with two different TSHβ gene mutations. We also reviewed published data on patients having the c.Q49X TSHβ gene mutation.
Materials and Methods
Case descriptions
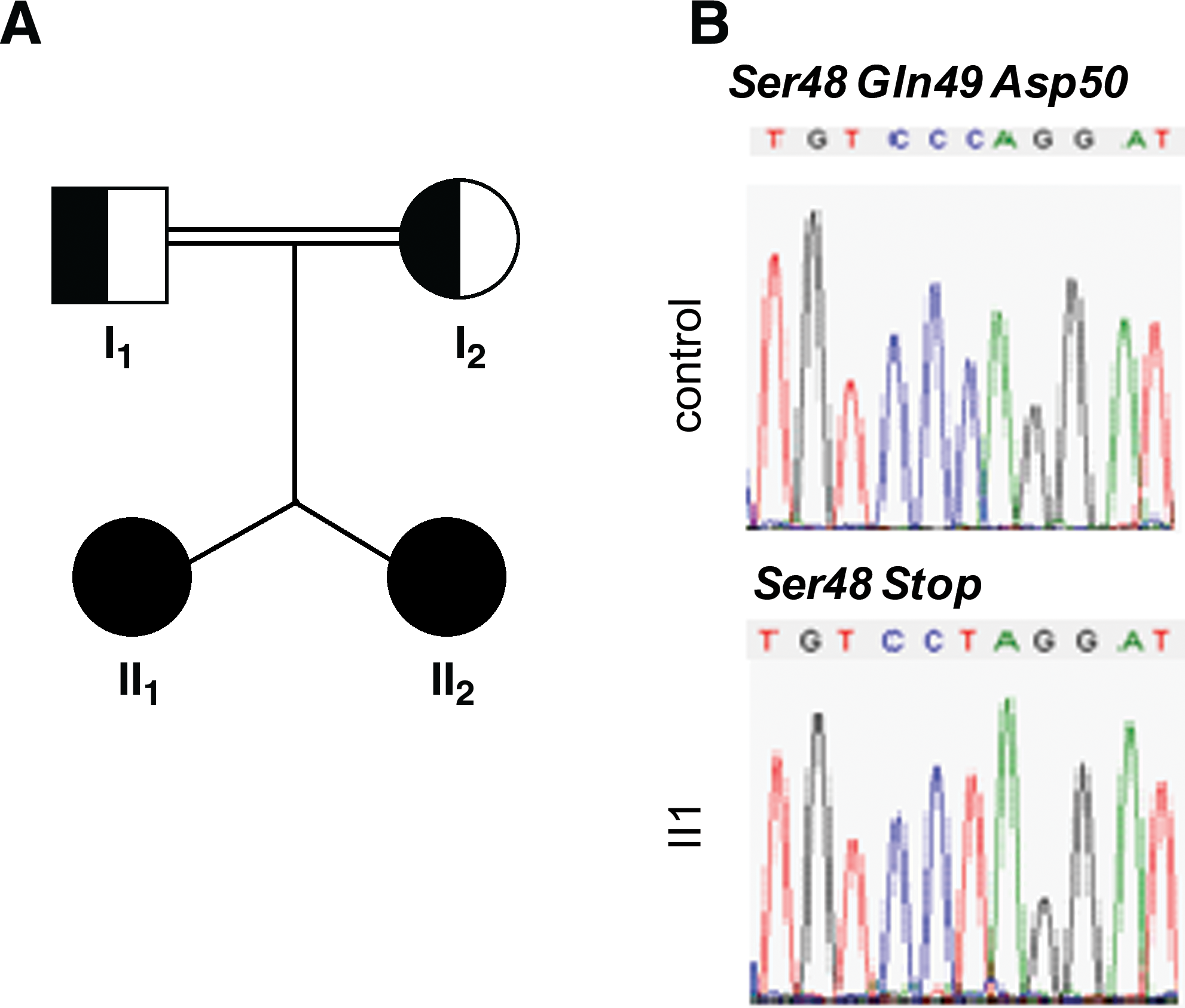
Family of Greek/Armenian descent (Fig. 1A)
(
The first family included two monozygotic twin sisters (patients II-1 and II-2) of consanguineous parents who were referred at 10.5 months of age for feeding problems, constipation, excessive sleep, low activity, and delays in growth and neuromotor development (Table 1; see also Supplemental Table S1, available online at
See Bonomi et al. (16).
See Vuissoz et al. (17).
See Sertedaki et al. (18).
The present study.
Respiratory distress in patient 5 required intubation.
Confirmatory TSH at neonatal screening (chemiluminescent immunoassay; Beckman, Fullerton, CA).
NA, not available; RD, respiratory distress; HG, hypoglycemia; TSH, thyrotropin.
See Bonomi et al. (16).
See Vuissoz et al. (17).
See Sertedaki et al. (18).
The present study.
At diagnosis.
Peak value after thyrotropin-releasing hormone stimulation.
Normal pituitary gland.
T4, thyroxine; US, ultrasound; MRI, magnetic resonance imaging.
At 10.5 months of age, the twins were started on levothyroxine (LT4) replacement therapy. The circulating fT4 and triiodothyronine (T3) levels returned rapidly to the normal range, indicating good adherence to the treatment regimen. The physical and mental development of both twins improved, and the typical signs of hypothyroidism resolved. At last follow-up, the twins were 2.4 years of age and were taking a maintenance LT4 dose of 45 μg/day (5 μg/[kg · day]). Their heights were 81 cm (−1.5 SD) and 81.8 cm (−1.2 SD), respectively, indicating catch-up growth. Both parents had normal thyroid function tests and normal TRH test results (data not shown).
Family of Polish descent (Fig. 2A)

(
This family of Polish descent was referred to us for genetic testing after the birth of the third affected child. The oldest sibling (patient II-1) had a TSH level of 0.04 mIU/L at neonatal screening but was diagnosed with hypothyroidism only at 2 months of age when he was evaluated for persistent jaundice, hypotonia, and constipation (Table 1 and Supplemental Table S1). Thyroid function tests showed an fT4 level of 4.24 pmol/L and a TSH level of 0.03 mIU/L. When he was 12 years and 9 months of age, a TRH test performed after 3 weeks without LT4 confirmed the diagnosis of CCH by showing low TSH levels of 0.06, 0.04, 0.02, 0.02, and 0.01 mIU/L at 30, 60, 90, 120, and 180 minutes after TRH, respectively (basal fT4, 1.54 pmol/L; basal T3, 0.012 nmol/L; and basal TSH, 0.02 mIU/L) (Table 2). Moreover, the basal prolactin level was normal (8 μg/L), as well as the prolactin response to TRH (10 μg/kg body mass) (51, 33, 22, 16, and 11 μg/L at 30, 60, 90, 120, and 180 minutes, respectively). At last follow-up, he was 12 years and 9 months of age and was taking LT4 in a dosage of 75 μg/day (1.8 μg/[kg · day]). His height was 155 cm (+0.5 SD; target height, 185 cm, +2.2 SD).
The second child was a girl (patient II-2) who was 3 years younger than patient II-1. She was first evaluated at 1 month of age for jaundice and constipation. Her fT4 level was <2.5 pmol/L and her TSH level was 1.9 mIU/L, establishing the diagnosis of CCH. At last follow-up, she was 10 years of age with a height of 139.5 cm (+1 SD; target height, +2 SD) and was taking an LT4 dosage of 62.5 μg/day (2.4 μg/[kg · day]).
In the index case, who is the youngest sister (patient II-3), the family history prompted tests at birth, which showed an fT4 level of 2.5 pmol/L and a TSH level of 0.1 mIU/L, indicating CCH. LT4 therapy was started on the first postnatal day. At last follow-up, she was 7 years of age with a height of 124 cm (+1.2 SD; target height +2 SD) and was taking an LT4 dosage of 50 μg/day (2.5 μg/[kg · day]).
Retrospectively, we found that TSH levels at neonatal screening were very low in all three affected siblings (0.04 mIU/L in the oldest sibling, 0.01 mIU/L in the middle sibling, and 0.1 mIU/L in the index patient). In all three patients, serum fT4 levels were measured using a direct back-titration method based on DELFIA® Technology (Wallac, Turku, Finland) and other hormones were assayed using sensitive and specific commercial kits. MRI of the brain was performed at 7 years of age in the oldest patient and at 2 years of age in each of the other two siblings, which showed normal findings in all three patients.
Genotype analysis
Genomic DNA was extracted from whole blood using standard techniques in the five above-described patients and in their parents. The coding region of the TSHβ gene, including exon/intron boundaries, was amplified by polymerase chain reaction (PCR), using the previously described primers and conditions (19). PCR products were purified using the Qiaquick PCR purification kit (Qiagen, Hilden, Germany) and sequenced using the ABI PRISM Dye Terminator cycle sequencing Ready Reaction kit (PE Applied Biosystems, Foster City, CA) according to the manufacturers' instructions. Sequences were analyzed using Sequence Navigator Software (PE Applied Biosystems). Bidirectional sequencing was performed using an automated cycle sequencer (ABI3100 genetic analyzer; PE Applied Biosystems). Sequence alterations were examined in the context of the open reading frame to determine whether the alteration changed the corresponding amino acid.
Review of the literature on the C.Q49X TSHβ gene mutation
We performed a systematic review of the literature by searching MEDLINE using the key words “congenital hypothyroidism,” “central hypothyroidism,” “TSHβ gene,” and “mutation.” We included articles about patients with a documented mutation analysis for whom individual clinical data were available. We reviewed data on all patients with the c.Q49X TSHβ mutation, and we recorded the age at CCH diagnosis, laboratory test findings, imaging study findings, result of neonatal screening, TSH, T4, and T3 levels, thyroid ultrasound, and cerebral MRI. We identified five patients from three families reported in three papers until December 10, 2009 (16 –18). Data on patients harboring the c.C105Vfs114X mutation, the most frequent TSHβ mutation, were reviewed recently (11,20).
Results
Sequencing and genotyping
Family of Greek/Armenian descent
Direct sequencing of exons 2 and 3 of the TSHβ gene in both patients and their parents showed that both twins were homozygous for a substitution of cytidine for thymidine at nucleotide position 145 (g.145C > T), thereby replacing glutamine (CAG) by a premature stop codon 49 (c.Q49X). Both of the euthyroid consanguineous parents were heterozygous for this mutation (data not shown) (Fig. 1B).
Family of Polish descent
The electropherogram showed a homozygous T-nucleotide deletion at codon 105 of the TSHβ gene in the index patient and a heterozygous mutation in the euthyroid parents (data not shown). The base pair deletion (g.313delT) causes a frame shift with substitution of a cysteine codon (TGT) for a valine codon (GTA) in the new reading frame (c.C105V) and generation of a premature stop at codon 114 (114X) (Fig. 2B).
Review of the literature on the C.Q49X mutation
As shown in Tables 1 and 2 and Supplemental Table S1, five patients with the c.Q49X TSHβ mutation from three different families have been previously reported (16 –18). Thus, with our two patients from the Greek/Armenian family, we had data on seven patients. Consanguinity was a feature in three of the four families. The patients varied considerably in terms of the CCH phenotype at diagnosis and of the other features such as birth weight/intrauterine growth and gestational age at birth (Tables 1 and 2 and Supplemental Table S1). The diagnosis was established early in three patients (no. 5, 6, and 7) who had severe perinatal manifestations and late in the other four patients upon evaluation of severe clinical manifestations of hypothyroidism. Serum TSH levels also varied widely across patients, from undetectable to normal, but differences in assay systems should be considered; however, the TRH test consistently showed an impaired or absent TSH response, establishing the diagnosis of CCH. During TRH testing, the patients typically exhibited either an abnormally small increase or a slightly delayed but excessive increase followed by a delayed decrease in plasma TSH concentrations (Table 2).
Discussion
To date, TSHβ gene mutations are the only known cause of TSH deficiency. Seven mutations have been described in 50 patients with CCH belonging to 34 families (11).
We describe five new patients with two different, previously described, homozygous TSHβ mutations (c.Q49X and c.C105Vfs114X) responsible for isolated CCH. The diagnosis was made late in childhood, except for the youngest sibling of the second family, in whom the family history led to early testing. In agreement with previous reports, the index cases in our families had normal neonatal screening results followed by severe, early-onset, nonautoimmune hypothyroidism. Thus, the diagnosis was suspected only when clinical symptoms of hypothyroidism developed. Nevertheless, the undetectable TSH levels in the three affected siblings of the Polish family suggest that very low TSH levels at neonatal screening may deserve attention. We are not aware of studies addressing this issue. Further studies of patients with CCH are needed to determine whether the poorer outcomes associated with CCH are related to the older age at treatment initiation or to the lower hormone levels at diagnosis and whether the diagnosis can be made earlier using neonatal TSH screening only.
Both our patients with the c.Q49X mutation had mild hypoglycemia, despite normal levels of cortisol and GH. This feature may be related in part to the major role in neonatal lipolysis played by TSH released upon activation of the hypothalamic–pituitary–thyroid axis (21,22). Indeed, alteration in the hepatic metabolism and defects in the lypolytic enzyme activities could be related to CH (23,24). Therefore, in addition to hormone replacement therapy, CCH related to TSHβ gene mutations may require other interventions targeting possible neonatal complications such as hypoglycemia.
In our five patients, ultrasonography showed hypoplasia of the thyroid gland. Although TSH is not required for the development and migration of the thyroid anlage, it is essential for thyroid growth and function in the postnatal period. Thus, TSH deficiency or inactivity typically results in a thyroid gland that is normally located but hypoplastic and hypofunctional (25,26). However, goiter was also found in three of six adults with isolated TSH deficiency (27).
A review of the literature allowed us to identify five additional patients with the c.Q49X mutation. The recessive inheritance in a consanguineous setting in most of the reported cases is consistent with either common ancestry or de novo mutations in a hot spot (16 –18). The nonsense c.Q49X mutation in exon 3 of the TSHβ gene leads to the formation of a truncated TSH β-subunit that is less than half as long as the normal mature peptide. Indeed, the TSHβ seat-belt region (aa 88–105) is necessary for proper dimerization with the α-subunit and subsequent secretion of a mature TSH heterodimer. Therefore, the c.Q49X TSH β- and α-subunits may be unable to form stable heterodimers. The result would be deficient TSH secretion, as TSH is secreted only after the α- and β-subunits become linked (16,18). Further, the C-terminus of the TSH β-subunit, which is absent in patients having the c.Q49X mutation, also contains domains required for binding with the TSH receptor. The c.C105Vfs114X mutation results in loss of seat-belt stability due to disruption of one of the two cysteines that are important for disulfide bond formation. The mutant protein may not be secreted and/or may have reduced biological activity (28).
Considerable variability in TSH levels exists across patients with the c.Q49X mutation, some of whom have normal or high circulating TSH levels measured using a monoclonal or polyclonal antibody assay (Table 2) (29). This observation may be ascribable in part to differences in assay methods and/or to the differences in TSH immunoreactivity and bioactivity that are common among CCH patients (Table 2) (3). Indeed, when assayed using systems that have limited specificity, TSH levels may be normal or even moderately elevated initially. Testing with recent immunoassays, however, shows very low or undetectable TSH levels. Therefore, the presence of immunoreactive but biologically inactive TSH raises diagnostic challenges (Table 1).
The phenotypic variability seen even among family members carrying the same mutation suggests genetic and/or nongenetic background. The regulation of thyroid growth and function by the hypothalamic–pituitary axis may change after birth, and this change may explain the improvement in thyroid function seen over the first few years of life in patients with congenital thyroid hypoplasia (30). Epigenetic modifications and variations in the expression or modulation of genes in the surrounding parenchyma and developing blood vessels that supply the thyroid gland may be involved (31). In addition, changes in environmental factors such as adhesion molecules and vascular factors may play a role (32).
The second mutation described here affects codon 105 (TGT) of the TSHβ gene, where the thymine at the third base position is deleted, producing a reading frame shift and a downstream premature termination signal (c.C105fs114X). This mutation has been reported chiefly in Western Europe but also in other continents and is the most common mutation known to occur in patients with isolated CCH. None of the parents identified so far were consanguineous, suggesting a de novo mutation in a hot spot (11). However, a founder effect might be involved in some cases (33). A study of heterozygote carrier frequency in the general population of Germany suggests a low disease risk of about 10 newborns per 700,000 births per year (33). Codon 105 encodes a cysteine residue that is critical for TSH β-subunit dimerization with the α-subunit. Thus, the c.C105fs114X mutation leads to the production of a truncated protein that is unable to form dimers (34).
In conclusion, we describe two new sibships with isolated CCH due to two different homozygous TSHβ gene mutations (c.Q49X and c.C105fs114X). The prevalence of TSHβ gene mutations is unknown, and it is important to bear in mind that isolated TSH deficiency is missed by neonatal screening tests that are based on TSH measurement. Therefore, infants with a clinical suspicion of hypothyroidism should undergo a thorough evaluation, even when their TSH-based screening tests are normal. Very low levels of TSH at neonatal screening may cause the diagnosis to be suspected. In addition, phenotypic variability has been documented among patients with CCH, even those carrying the same mutation. Therefore, the TSHβ gene mutation alone may fail to fully explain the phenotypic variability, suggesting that other mechanisms modify the phenotype.
Footnotes
Acknowledgments
This work was supported in part by grants from Électricité de France (RB 200605) and the European Society for Paediatric Endocrinology Research Unit (to M.P.), and Sao Paulo State Research Foundation (FAPESP Grant 06/54950-6) (to R.M.B.M.). H.E.R. was supported by grants from Capes (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, BEX/Programa de Pós-Doutorado no Exterior) and “Ville de Paris.”
Disclosure Statement
The authors have no conflicts of interest to declare.
This work was presented in part at the European Thyroid Meeting, 2009, Lisbon, Portugal.