
Abstract
Background:
Approximately 5% of the nonmedullary thyroid cancers are hereditary. Hereditary nonmedullary thyroid cancer may occur as a minor component of familial cancer syndromes (familial adenomatous polyposis, Gardner's syndrome, Cowden's disease, Carney's complex type 1, Werner's syndrome, and papillary renal neoplasia) or as a primary feature (familial nonmedullary thyroid cancer [FNMTC]). The goal of this article was to review our current knowledge on the hereditary nonmedullary thyroid cancer.
Summary:
Epidemiologic and clinical kindred studies have demonstrated that FNMTC is a unique clinical entity. Most studies suggest that FNMTC is associated with more aggressive disease than sporadic cases, with higher rates of multicentric tumors, lymph node metastasis, extrathyroidal invasion, and shorter disease-free survival. A hereditary predisposition to nonmedullary thyroid cancer is well established, but the susceptibility genes for isolated FNMTC have not been identified. However, additional susceptibility loci for FNMTC have been recently identified in classic isolated cases of FNMTC (1q21, 6q22, 8p23.1-p22, and 8q24).
Conclusions:
More studies are needed to validate chromosomal susceptibility loci and identify the susceptibility genes for FNMTC. The discovery of the predisposing genes may allow for screening and early diagnosis, which could lead to improved outcomes for patients and their families.
Introduction
Thyroid cancers of follicular cell origin account for more than 95% of all the thyroid cancer cases, and the remaining cancers originate from the parafollicular cells (medullary). Thyroid cancers of follicular cell origin are classified into four groups: papillary, follicular, Hürthle cell, and anaplastic (6). Although medullary thyroid cancer (MTC) accounts for 5% of all the thyroid cancers, it is responsible for about 15% of all the deaths related to thyroid cancer (7 –12). Hereditary medullary thyroid carcinoma (HMTC) encompasses about 25% of all the medullary thyroid carcinomas. It may present as a component of multiple endocrine neoplasia type 2 (MEN2) syndrome or as isolated MTC (familial MTC).
Activating, germline mutations in the RET (tyrosine kinase receptor, rearranged during transfection) proto-oncogene, located on chromosome 10q11.2, are responsible for HMTC (7,13,14). Before genetic testing for germline RET mutation was developed in 1993, basal and stimulated blood calcitonin tests were used to screen at-risk family members of patients with MTC. Almost all patients with a germline RET mutation develop c-cell hyperplasia or MTC during their lifetime. Today, about 98% of all the mutations responsible for HMTC are known. Somatic RET point mutations have been identified in 40–60% of the patients with sporadic MTC (15). All patients from families with HMTC with a confirmed RET mutation should undergo prophylactic total thyroidectomy, with or without central neck dissection, depending on whether MTC is present (16). The timing of prophylactic surgery should be based on the RET mutation genotype, as there is evidence that a genotype–phenotype association exists (17,18).
Hereditary nonmedullary thyroid cancer (HNMTC) may occur as a minor component of other familial cancer syndromes (familial adenomatous polyposis (FAP), Gardner's syndrome, Cowden's disease, Carney's complex type 1, Werner's syndrome, and papillary renal neoplasia) or as a primary feature (familial nonmedullary thyroid cancer [FNMTC]). The relative frequency of different forms of familial thyroid cancer is shown in Figure 1 (19). The susceptibility genes for FNMTC, such as the germline RET mutations for HMTC, has not yet been identified. Such genes could be of great value to screen at-risk individuals, thereby making early diagnosis and prophylactic thyroidectomy possible, as well as selection of appropriate adjuvant therapy, which could lead to better outcomes.
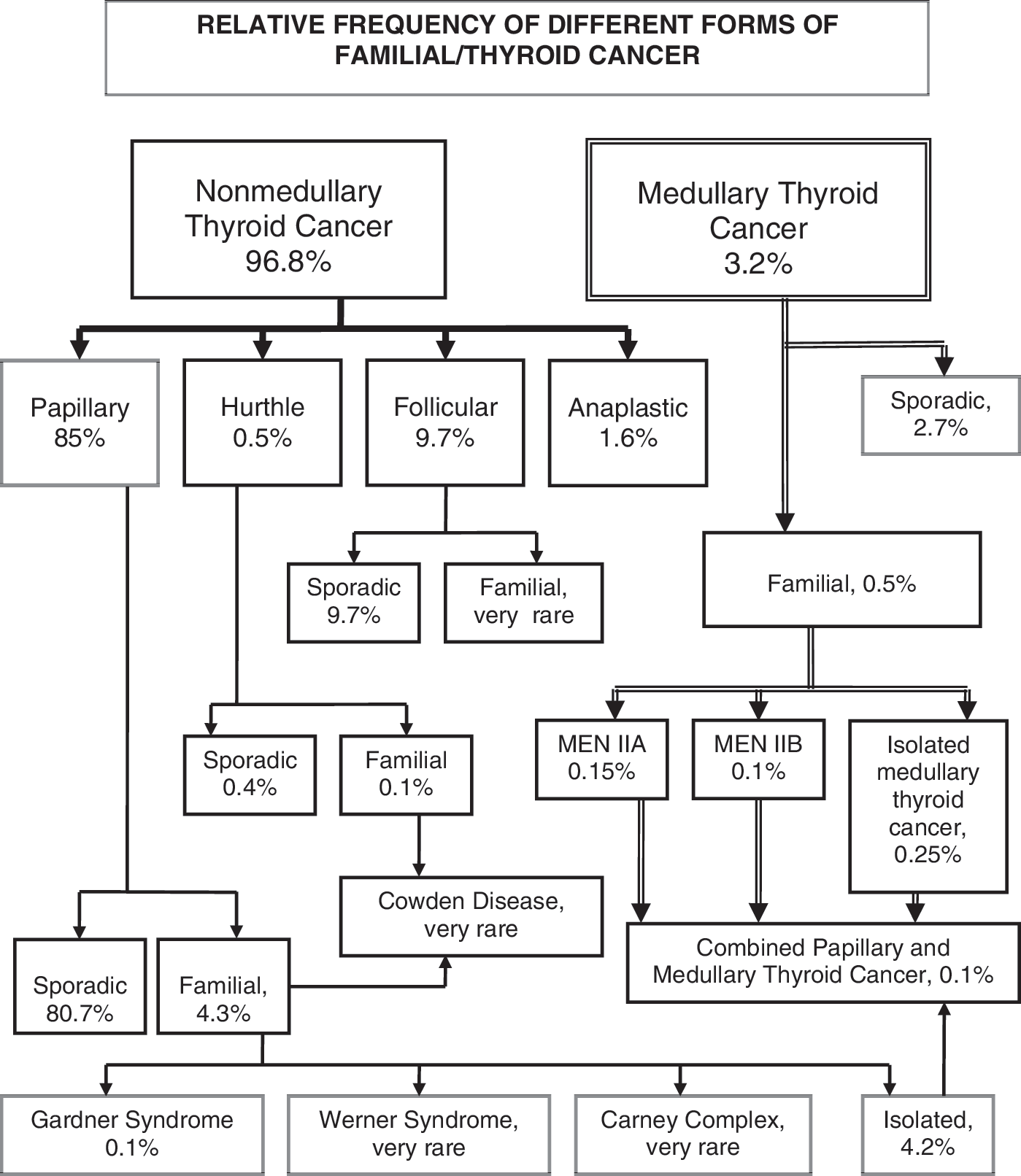
Relative frequency of the different forms of familial/thyroid cancer. Data adapted from the National Cancer Database; Surveillance, Epidemiology and End Results registry of the National Cancer Institute; and the Endocrine Surgery data bank at the University of California, San Francisco, CA.
Familial Nonmedullary Thyroid Cancer
FNMTC accounts for 3.2–6.2% of all the thyroid cancer cases and has an autosomal dominant pattern of inheritance with incomplete penetrance. FNMTC is defined by the presence of well-differentiated thyroid cancer of follicular cell origin in two or more first-degree relatives, in the absence of other predisposing hereditary or environmental causes. Charkes (20) estimates that the disease may actually be sporadic in up to 69% of the families with a family history of only two affected members, as opposed to 1–4% in those with three or more affected members.
PTC is the most common histological subtype of FNMTC, although kindreds with follicular and Hürthle cell subtypes of thyroid cancer have also been reported. FNMTC occurs at an earlier age, especially in nonindex family members, which may be because of a lead-time bias in the diagnosis of successive generations. FNMTC behaves slightly more aggressively than its sporadic counterpart, leading many medical centers to recommend more aggressive management of the affected family members (21 –26).
HNMTC may occur as a minor component of familial cancer syndromes (FAP, Gardner's syndrome, Cowden's disease, Carney's complex type 1, Werner's syndrome, and papillary renal neoplasia) or as the predominant feature in FNMTC. The clinical characteristics and genetic causes of these syndromes are shown in Table 1 (27 –38). The following syndromes are possibly associated with the development of nonmedullary thyroid cancer, but the link is much less established than in the aforementioned examples: McCune–Albright syndrome, Peutz–Jeghers syndrome, and Ataxia-teleangiectasia (Louis–Bar's syndrome). Although many of the susceptibility genes for HNMTC associated with familial cancer syndromes are known, these cases account for only a small number of cases (Fig. 1).
FAP, familial adenomatous polyposis; PTC, papillary thyroid cancer; GI, gastrointestinal; FTC, follicular thyroid cancer; ATC, anaplastic thyroid cancer.
Clinical Features, Treatment, and Outcome of HNMTC
Most studies, including large cohort studies in the past decade, suggest that HNMTC is more aggressive than sporadic differentiated thyroid cancer. HNMTC is associated with a higher rate of multicentric tumors, coexistent benign thyroid tumors, extrathyroidal invasion, and lymph node metastasis. One of these large cohort studies had a multicenter case-matched design (24) and found a higher rate of recurrence in patients with HNMTC. Another had a double-cohort design (26) and found significantly higher rates of multicentric tumors, lymph node metastasis, and benign thyroid tumor, but no difference in age at diagnosis, primary tumor size, rate of extrathyroidal invasion, and distant metastasis. Perhaps the most compelling study demonstrating that HNMTC may be more aggressive was a descriptive study of 119 patients with papillary thyroid microcarcinoma (39). Seven of these patients had HNMTC, and the rates of tumor multicentricity, bilobar disease, lymph node metastasis, vascular invasion, and recurrence rates were all significantly higher than in sporadic cases. Finally, in a study of 47 parents and children with HNMTC (23) relationship, HNMTC displayed the features of clinical anticipation with the second generation acquiring the disease at an earlier age and having more advanced disease at presentation. Therefore, a molecular mechanism for anticipation for HNMTC has been suggested as occurs in other hereditary diseases such as Huntington disease because of an unstable trinucleotide repeat expansion and for congential dyscheratosis because of telomerase RNA component mutation (23). Although some investigators have observed no difference in the clinical features of HNMTC as compared with the sporadic cases, most studies indicate an aggressive disease phenotype.
Fine-needle aspiration (FNA) cytology is a cost-effective and reliable diagnostic test for thyroid nodules. It accurately diagnoses most cases of PTC, but may have a lower sensitivity in cases of HNMTC. In a previous study by our group (40), three clinically significant tumors were missed by the FNA cytology (12%) when the tumors were found outside the index nodule. Two of these patients with false-negative FNA results had locoregional spread at the time of surgery and one patient had three recurrences. Taken together, these findings suggest that HNMTC is more aggressive than sporadic disease, but no difference in survival rate has been reported. Because thyroid cancer has a relatively low mortality rate, it is unlikely that any study would have a large enough cohort with a long enough follow-up period to detect a small difference in survival. Several investigators have, however, observed that the disease-free survival in patients with HNMTC is lower than in patients with sporadic disease, especially among those with three or more affected family members (24,26,41). Moreover, when life expectancy is used as an endpoint in comparison to the unaffected family members, life expectancy is lower for HNMTC when three or more cases are present (41). We therefore recommend total thyroidectomy with a prophylactic central neck lymph node dissection if the patient has a malignant thyroid nodule and a strong family history of thyroid cancer. If lymph node involvement is present in the lateral neck compartment by preoperative imaging or by palpation, a therapeutic lymph node dissection should also be performed.
Genetics of HNMTC
Syndromic HNMTC
HNMTC encompasses thyroid cancer cases associated with other hereditary cancer syndromes and cases of isolated nonmedullary thyroid cancer (FNMTC). Although the susceptibility genes for many of the hereditary cancer syndromes with thyroid cancer are known, these cases only account for a small subset of HNMTC cases.
Familial adenomatous polyposis
FAP is an autosomal dominant disease caused by inactivating mutations of the APC tumor suppressor gene located on chromosome 5q21. It is characterized by the development of multiple adenomatous polyps with malignant potential lining the mucosa of the gastrointestinal tract, particularly the colon, with an early age of onset. FAP occurs in several autosomal dominant diseases, including Gardner's syndrome, Peutz–Jeghers syndrome, and Turcot's syndrome. PTC occurs in some families with FAP. These thyroid cancers have a unique cribriform pattern on histologic examination and occur more commonly at a young age (below 30 years) and in women. Most female patients with FAP and PTC also have a RET/PTC somatic mutation as well as the APC germline mutation (42).
Gardner's syndrome
In this variant of FAP, polyposis of the large bowel is associated with the extracolonic manifestations of supernumerary teeth, fibrous dysplasia of the skull, osteomas of the mandible, fibromas, desmoid tumors, epithelial cysts, hypertrophic retinal pigment epithelium, upper gastrointestinal tract hamartomas, hepatoblastomas, and thyroid tumors. Thyroid neoplasms occur at a young age (below 30 years) and in women. The overall risk of developing thyroid cancer is ∼2%.
Cowden's disease (multiple hamartoma syndrome)
This autosomal dominant disorder is associated with a mutation in the PTEN tumor suppressor gene on chromosome 10q22-23. It is characterized by hamartomas and other tumors of the thyroid, breast, colon, endometrium, and brain. The most frequent extracutaneous manifestations of this disease are thyroid neoplasms, both benign neoplasms and follicular thyroid cancers, occurring in approximately two-thirds of the subjects.
Werner's syndrome (adult progeria)
This autosomal recessive disease is characterized by premature aging, scleroderma-like skin changes, cataracts, subcutaneous calcifications, muscular atrophy, diabetes, and a high incidence of neoplasms. It has been linked to mutations of the WRN gene on chromosome 8p11-21. Thyroid cancer occurs at a young age and is predominantly follicular. Papillary and anaplastic cancers are also more common.
Carney's complex
This autosomal dominant disease is caused by the mutation of the PRKAR1 gene on chromosome 17q24. It is characterized by myxomas of the soft tissues, skin and mucosal pigmentation (blue nevi), schwannomas, and tumors of the adrenal gland, pituitary, and testicle. Thyroid gland disease is common (∼11% of the patients) and includes adenomatous hyperplasia, cysts, or PTC or follicular thyroid carcinoma.
Papillary renal neoplasia
Malchoff et al. (43) mapped the PRN1 locus on chromosome 1q21 in an American family with the combination of PTCs, benign thyroid nodules, and papillary renal neoplasia.
Nonsyndromic HNMTC
On a molecular level, the genetic foundations of FNMTC as a distinct syndrome remain poorly understood (Table 2). Unlike in the case of HMTC (MEN2A, MEN2B, and familial medullar thyroid cancer) syndrome, which is because of the germline point mutations in the RET proto-oncogene (44), the causative genes predisposing to FNMTC have not been identified. Several groups, including our own, have attempted to identify chromosomal loci associated with FNMTC using linkage analysis. The first study by the French NMTC Consortium identified the thyroid cancer with oxyphilia (TCO) locus on chromosome 19p13.2 in a French family with oxyphilic thyroid tumors (45). An Australian group identified NMTC1 on 2q21 in a Tasmanian kindred with follicular variants of PTC (46). Attempts to validate these results have met with varying success. In particular, the TCO and NMTC1 loci have been confirmed in a few independent families, and there is some evidence to suggest that the two loci may in fact interact with one another, further increasing the risk of thyroid cancer. Novel variants of TIMM44, a gene that maps to the TCO locus, appear to cosegregate with the TCO phenotype, although in vitro functional experiments suggest that they do not lead to the loss-of-function effects reminiscent of a tumor suppressor gene (47). Recently, the loss of heterozygosity at the TCO and NMTC1 loci was demonstrated in some, but not all tumor specimens from patients with HNMTC (48). These findings suggest that alterations at TCO and NMTC1 are important in a fraction of cases with HNMTC. This is not surprising as most cases of FNMTC are PTC and rarely TCO (Hürthle cell) or follicular variant of PTC.
PRN, papillary renal neoplasia; TCO, thyroid cancer with oxyphilia.
Prazeres and colleagues (49) reported the finding of loss of heterozygosity at the 19p13.2 and 2q21 loci in tumors from familial clusters of NMTC, providing evidence that the inactivation of putative genes in these regions, acting as tumor suppressors, may be involved in the development of tumors in the context of HNMTC. In contrast, the de novo linkage analysis studies on HNMTC family cohorts have shown low or no linkage to suggest the presence of chromosomal susceptibility loci. These conflicting results underscore the main limitations of the positive studies mentioned above, namely that they were only characterized in one or a few affected families, and all but one examined rare variants of HNMTC. Another important shortcoming of many previous studies is that a less stringent definition for FNMTC may have diluted the linkage studies with sporadic cases. It is also possible that FNMTC may be a heterogeneous inherited syndrome with more than one susceptibility gene with somatic mutations that alter disease initiation and progression.
Other molecular approaches and techniques have been used to identify and characterize genetic alterations associated with FNMTC. One of these approaches is the comparative genomic hybridization, which provides a global analysis of the chromosomal changes (50). However, it has failed to reveal any FNMTC-specific chromosomal gains or losses (51). Multiple germline mutation analyses have excluded the most common somatic mutations in genes associated with sporadic thyroid cancers—including BRAF, RET, RET/PTC, MET, MEK1, MEK2, RAS, and NTRK—as the candidate genes for HNMTC (52). Even though no germline mutations were found, a similar prevalence and distribution in the common somatic mutations (RAS/BRAF) were observed by Cavaco et al. (48). The investigators suggest that these somatic genetic alterations may be involved in the FNMTC tumor progression. Using newer, higher genomic resolution techniques such as the single-nucleotide polymorphism (SNP) array platforms, a Portuguese group recently identified a locus on 8p23.1-p22 in a Portuguese family with a more classic FNMTC phenotype (53). He et al. (54) also recently identified 8q24 as a susceptibility locus for PTC in a large family with PTC and melanoma. This was validated in 25 additional families with PTC. This locus had two known genes (thyroglobulin and Src-like adaptor), which on sequencing, however, showed no mutations in the coding or regulatory regions. But analyses of the candidate noncoding RNA genes showed that AK023948 was a possible candidate gene because it was significantly downregulated in the majority of PTC. Our group recently attempted to address these limitations by performing a SNP array-based linkage analysis of 38 families with classic FNMTC pedigrees; we believe that pooling a broad sampling of families best addressed the question of whether linkage loci truly exist in most HNMTC cases (45 PTCs, 1 follicular variant of PTC, and 3 follicular thyroid cancers). Our preliminary results have shown significant linkage loci at 1q21 and 6q22, and further efforts to validate these results prospectively in other families are under way, as well as to identify the candidate genes.
Recently, a large genome-wide association study was undertaken in Iceland, comparing the SNP array profiles of 192 people with PTCs and follicular thyroid cancers to those of >37,000 healthy controls, which revealed strong association signals at markers on 9q22.33 and 14q13.3 (55). Although these results suggest the presence of certain genotypes that predispose to thyroid cancer, it is unclear whether these population-based genome-wide association study results can be extrapolated to the narrower cohort of families with FNMTC, which may or may not overlap.
MicroRNA (miRNA) has recently been implicated as an important contributor to carcinogenesis. Jazdzewski et al. (56,57) showed that among 608 patients with PTC, a common polymorphism in pre-miR-146a affected the miRNA expression, which contributed to the genetic predisposition to PTC. To our knowledge, no studies have focused specifically on FNMTC to determine if the miRNA expression profiles between sporadic and FNMTC are different.
Several investigators using the genome-wide microarray analysis have shown that the classification of the nonmedullary thyroid tumors could be refined. For example, Fontaine et al. (58) recently demonstrated the gene expression levels that classify the nonmedullary thyroid lesions as either benign or malignant. However, no studies have been done to determine if the genome-wide RNA expression profiles between sporadic and FNMTC are distinct from each other. Finally, although several proteomic analyses have been developed to identify the potential markers of thyroid cancer, no distinct proteomic profile has been reported for HNMTC in particular (59,60).
Conclusions
HMNTC may occur as a component of other familial cancer syndromes or as isolated cases. FNMTC has been established by epidemiologic and kindred studies as a distinct familial syndrome that may be associated with more aggressive behavior, and may warrant more aggressive treatment. The susceptibility genes for FNMTC have not yet been identified. There is likely to be more than one susceptible gene, given the nonuniform criteria used to establish the hereditary nature of HNMTC in most studies and the heterogeneous group of the histologic variants. A multicenter analysis that focuses on stringent criteria could lead to the identification of the susceptibility genes, which could lead to screening and early diagnosis of FNMTC, prophylactic treatment, and improved patient outcome.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.