
Abstract
Background:
Thyroid cancer is the most common endocrine tumor and is increasing in incidence. The aim of this study was to review mouse models of differentiated thyroid cancer and how they elucidate human thyroid cancer biology.
Summary:
Differentiated thyroid cancer, primarily papillary and follicular, comprises the majority of thyroid cancers. There has been tremendous growth in the cross-talk between basic science and clinical practice for thyroid cancer management. Insight into the framework of genes responsible for differentiated thyroid cancer has been gained through the use of mouse models. Common genetic alterations found in human papillary thyroid cancer such as RET/PTC rearrangements or the BRAFV600E mutation have genetically modified mouse counterparts. These and other preclinical mouse models have validated the importance of the cyclic adenosine monophosphate (cAMP)/protein kinase A and mitogen-activated protein kinase (MAPK) signaling pathways in papillary thyroid cancer (PTC). RAS mutations have a role in both papillary and follicular thyroid cancer development. Mice with overactivation of the phosphatidylinol-3-kinase (PI3K)–AKT and/or thyrotropin-regulated signaling pathways have been found to develop follicular thyroid cancer. Additional mouse models of thyroid cancer that utilize inducible expression systems are in development or are being characterized and will better reflect the majority of human thyroid cancers which are non-hereditary. Advances in in vivo imaging of mice allow for earlier detection of metastasis and the ability to follow tumor growth or regression which may be used in evaluation of pharmaceutical agents.
Conclusions:
Mouse models have expanded our understanding of the altered signaling pathways that contribute to thyroid cancer tumorigenesis and provide a powerful tool to develop novel diagnostic approaches and therapies.
Introduction
Standard therapeutic approach for differentiated thyroid cancer patients includes surgery and thyroid hormone suppressive therapy to reduce thyrotropin (TSH) levels, often followed by radioactive I-131 treatment, but few good options exist for metastatic disease (3). Recently, the class of targeted agents known as tyrosine kinase inhibitors (TKIs) has shown benefit for patients with advanced disease (5 –8). The new American Thyroid Association (ATA) guidelines on thyroid cancer management state that TKIs may be considered for patients not in a clinical trial and with progressive or symptomatic metastatic disease (9). The burgeoning clinical interest in TKIs and other targeted therapies is partly due to the improved knowledge regarding the molecular basis of thyroid cancer.
There is a wide spectrum of scientific approaches available to model thyroid cancer and each has its own unique limitations. A major challenge of using human thyroid cancer tissue is its limited availability to compare genetic, epigenetic, and/or proteomic profiles between normal and malignant tissue along with patient heterogeneity. Cultured thyroid cancer cell lines are useful in dissecting out signaling pathways and predicting tumor response to pharmaceutical agents. However, the basic functional unit of the thyroid, the three-dimensional thyroid follicle, is lost in monolayered cultures. In two-dimensional culture systems, thyrocytes may also lose normal functions such as TSH responsiveness (10). Culture systems with three-dimensional matrices or other growth factors can now recreate thyroid follicles in vitro and provide additional information on tumor–stromal interaction (11 –13). A more recent concern raised by Schweppe et al. is that many of the existing thyroid cell lines are redundant or are not thyroid in origin (14).
Mice are a fundamental tool for cancer investigators and provide insight into tumor behavior that cannot be replaced, at yet, by in vitro modeling. The mouse strain B6C3F1, frequently used in bioassay and carcinogenicity studies, has a very low incidence of spontaneous thyroid cancer development, which makes it difficult to incorporate in studies (15). There are three general approaches for inducing tumors: carcinogenic compounds, implantation of tumor cells via a subcutaneous or orthtotopic route in immunocompromised mice, or genetically engineered models. Chemical compounds inducing thyroid cancer are typically goitrogens which decrease thyroid hormone levels, leading to TSH elevation and goiter development followed by infrequent development of follicular neoplasms (16). Immunodeficient mouse strains used to study cultured human cell lines in vivo include the severe combined immunodeficient (SCID) and athymic (nude) strains. There are subtle differences in their immune systems which may affect tumor development as SCID mice can also tolerate human lymphocytes unlike nude mice (17). The tumor implantation site influences tumor behavior as studies on thyroid and nonthyroid cancer cell lines show that growth and metastasis are facilitated if implanted orthotopically (18,19). As several published studies of orthotopic models of thyroid cancer used cell lines that are now found to be derived from colon cancer or melanoma (14), one must be careful in the conclusions previously drawn about thyroid cancer biology. Orthotopic models of thyroid cancer have been shown to develop lymph node and pulmonary metastases, but not other sites of distant metastasis (20). To model bone metastasis, intracardiac or direct injection of tumor cells into bone is required (21). One caveat to determining whether a distant metastasis is present is that it may be limited by the investigator's motivation and resources to look for unusual sites of distant metastasis such as the liver or brain. Similar to the techniques that are now clinically used to detect human thyroid cancer, advances in microimaging such as high-frequency ultrasound, microcomputed tomography, and micro-positron emission tomography will improve tumor detection in mice (22). Recent development of an orthotopic model of anaplastic thyroid cancer with a cell line that stably expresses green fluorescent protein showed the feasibility of detecting thyroid cancer metastasis in lymph nodes and lung by fluorescence imaging (23). Sequential visualization of cell lines that express luminescent proteins in in vivo studies can be used in the future to quantify tumor progression and eventual response to therapeutic agents (17).
Genetically engineered models include the traditional transgenic mouse which is created by injecting DNA, including the gene of interest, into fertilized oocytes, resulting in random integration and unpredictable copy numbers of the foreign DNA into the mouse genome (24). Several transgenic mouse models of thyroid cancer contain the gene of interest under the expression of the thyroglobulin (Tg) promoter, allowing for thyroid-specific expression as shown in Table 1 (25 –27). Manipulation of embryonic stem cells via homologous recombination allows for targeted gene manipulation and is the principle behind both knockout mice, which typically have no functional protein expression of the target gene, and knock-in mice, which have the transgene inserted at a specific locus. As all cells of these knockout and knock-in models contain the transgene, these are not representative of the majority of thyroid cancer cases that result from somatic mutations (28). In addition, transgenic and knock-in models have much earlier expression of the oncogene compared with acquired human thyroid cancer and these mice may develop compensatory mechanisms, limiting the opportunity to study thyroid cancer development and progression (25,29). To circumvent these problems, recombinase-mediated gene modification (Cre/Lox) is now used to introduce a somatic mutation in specific tissues and/or at a specific time (24,30 –32). In one example of the Cre/Lox system, an exon of the gene of interest is modified by flanking it with two 34-bp loxP sequences (i.e., floxed). These mice are then crossed with a mouse strain that expresses the Cre recombinase, leading to deletion of the floxed sequence and gene inactivation (Fig. 1). Another model, described in more detail below, is the Rap1bG12V model wherein a floxed, constitutively active mutant Rap1b gene upstream of an inactive Rap1b under control of the bovine Tg promoter is crossed with a tamoxifen-inducible Cre mouse so that in the presence of tamoxifen, Rap1b is inactivated (Fig. 1) (33). These advances in genetically engineered mice allow for both spatial and temporal control of gene expression.
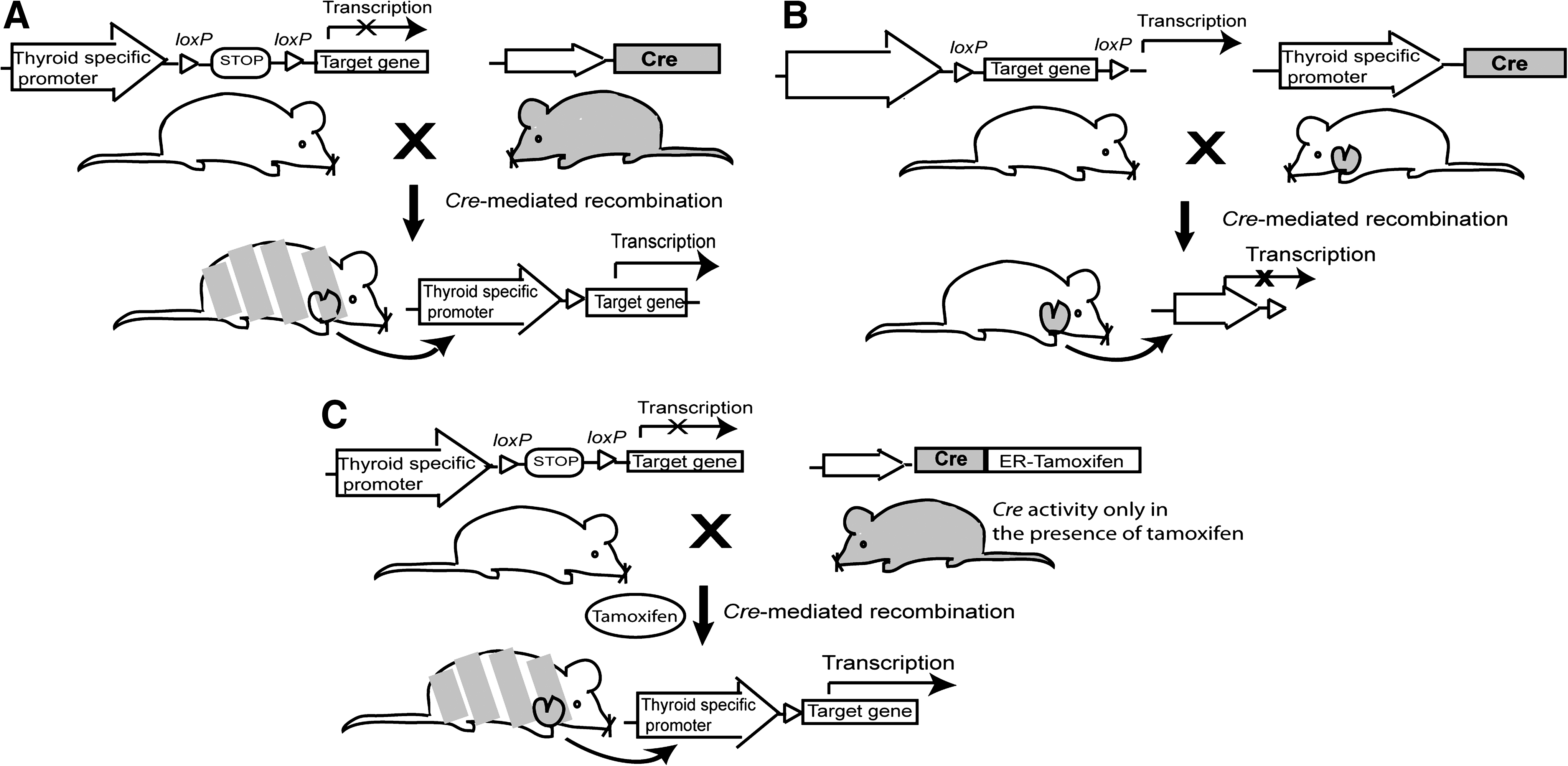
Examples of genetically engineered mice utilizing thyroid-specific promoters and Cre recombinase. (
RET, rearranged during transfection; PTC, papillary thyroid cancer; Tg, thyroglobulin; TRK-T1, neurotrophic tyrosine kinase receptor, type 1; TSH, thyrotropin.
Multiple variations of this approach have been designed for both oncogene activation and inactivation as exemplified in a lung cancer model (30). The sequence contains an oncogenic K-Ras mutation downstream of a floxed stop signal. Instead of crossing these mice with Cre mice, Cre-recombinase is delivered locally to the lungs via an adenovirus vector (30). When compared with the mouse model in which mutant Ras is expressed throughout the lung, this model recapitulates human lung cancer progression better, from a single individual somatic cell harboring an activated oncogene to macro disease (30).
Despite recent progresses, models generated from spontaneous mutations are rare (34). Another model that closely mimics sporadic somatic mutations uses endogenous intrachromosomal recombination mechanisms to simulate the random somatic activation of K-Ras. To make the chromosome susceptible for a mutagenic recombination event, two copies of exon 1 with one or two mutant alleles were inserted into the genomic site of the endogenous K-Ras allele in tandem. Intrachromosomal recombination between homologous sequences generates a single functional oncogenic K-Ras allele. In this model, all of these mice developed early onset lung adenocarcinomas (35).
The remainder of this review will focus on the genetically engineered models for differentiated thyroid cancer, specifically PTC and FTC. These models have enhanced our understanding of key signaling pathways central to thyroid carcinogenesis and can be used for future therapeutic agents.
PTC
The known genetic alterations found in PTC primarily affect two central signaling pathways in thyroid cells: TSH receptor (TSHR)-mediated signaling and mitogen-activated protein kinase (MAPK) pathways (Fig. 2). There is cross-talk between these pathways, and although mouse models to evaluate overactivation in either signaling pathways have been engineered, it is the models altering MAPK signaling which consistently recapitulate human PTC.
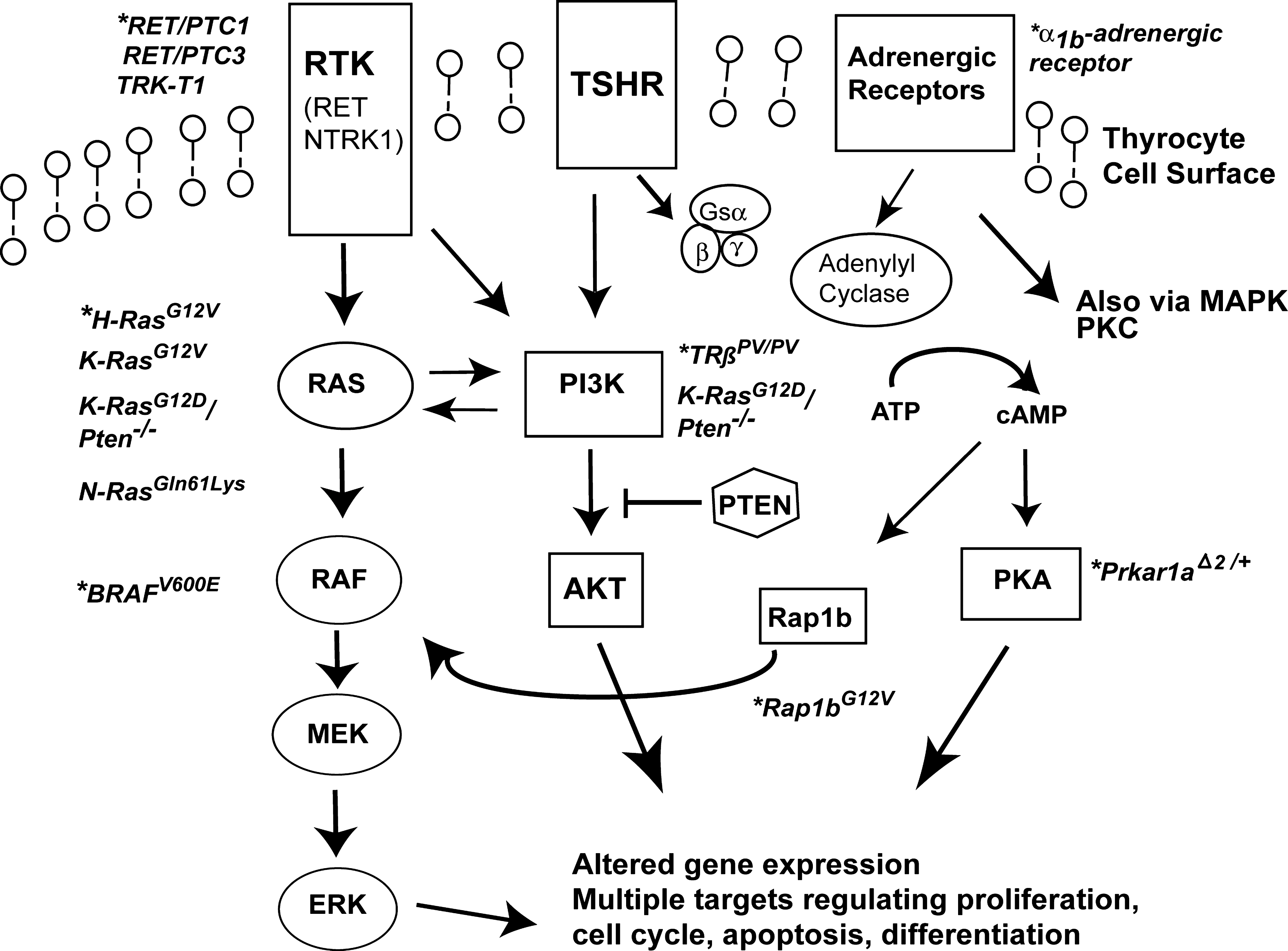
Altered signaling pathways and mouse models of thyroid cancer. Mouse models are indicated by an (*). Aberrant signaling through receptor tyrosine kinases (RTK) such as the RET and NTRK1 receptors induce papillary thyroid cancer (PTC). Overactivation of the RAS/RAF/MEK/ERK pathway (or MAPK signaling pathway) leads to PTC. The BRAFV600E mutation results in constitutive activation of BRAF and is commonly found in PTC. RAS overactivation is common to both papillary and follicular thyroid cancer (FTC) models. TSH receptor (TSHR) signaling via adenyl cyclases/cAMP/protein kinase A (PKA) is important for normal thyroid growth, but only a few models of TSHR overactivation develop thyroid cancer (Prkar1aΔ2/+, Rap1bG12V ). The adrenergic receptor may also contribute to thyroid cancer development also through increased cAMP and signaling via pathways such as MAPK and protein kinase C (PKC). Overactivation in the PI3K-AKT signaling pathway via cell-surface receptors such TSHR or inactivation of the phosphatase, PTEN, are important for FTC tumorigenesis. TRßPV/PV mice also have PI3K-AKT overactivation and develop FTC. Please see text for additional details regarding these mouse models.
TSHR and cAMP signaling
The TSHR is a classical G protein-coupled receptor and TSH binding to TSHR leads to activation of the heterotrimeric G protein (36). Then the stimulatory subunit Gsα dissociates from the G protein complex with eventual activation of adenylyl cyclases and generation of the secondary messenger, cyclic adenosine monophosphate (cAMP), leading to protein kinase A (PKA) signaling (37) (Fig. 2). The importance of this mitogenic pathway's role in thyroid cancer is the basis for thyroid hormone suppressive therapy in patients. However, it is unclear as to the importance of the TSHR/cAMP/PKA pathway in thyroid cancer initiation. Gsα mutations are only found in a small percentage of thyroid cancer, and a minority of patients with McCune-Albright syndrome who have upregulation of cAMP production develop thyroid cancer (38).
Mice made biochemically hypothyroid by antithyroid drugs have not reliably developed thyroid cancer despite dramatic TSH elevations. The adenosine A2 receptor also signals through G proteins and activation of cAMP/PKA signaling, but transgenic mice expressing the canine adenosine A2 receptor developed goiters and hyperthyroidism, but no thyroid cancer (39). Two other models which mimic TSHR overactivation via constitutive activation of Gsα include the Tg-gsp mouse which has a constitutively active Gsα subunit under control of the Tg promoter and a mouse that had thyroid-specific cholera toxin A1 subunit expression (40,41). Despite development of goiters and hyperthyroidism, these models did not develop thyroid cancer. A mouse model of PKA overactivation which recapitulates many findings of the human syndrome of PKA overactivation, Carney complex, was generated using heterozygous mice for a null allele of Prkar1a, the type 1a regulatory subunit of PKA. Only about 9% of these mice developed thyroid cancers, primarily identified as PTC; however, the earliest age-of-onset was 13.5 months (42). It appears that in mice, TSHR and/or PKA overactivation is not sufficient to initiate tumorigenesis but does enhance existing progression of PTC. Transgenic mice expressing the human papillomavirus E7 protein developed both PTC and FTC although this protein is not known to be important in human thyroid cancer (43). When these transgenic mice were crossed with the mice expressing the adenosine A2 receptor, these tumors became more aggressive, including development of metastasis (44). Additional work is required to understand the differences in human and mouse response to TSH/cAMP/PKA overactivation and thyroid cancer development.
RET/PTC mutations
Chromosomal translocations involving the proto-oncogene rearranged during transfection (RET) is one of the earliest identified genetic mutations in PTC (45). RET is normally not present in thyroid epithelial cells, but somatic RET/PTC mutations are commonly found in PTC, particularly radiation-induced thyroid cancer (37,46). RET is a tyrosine kinase receptor and chimeric RET/PTC proteins involve fusion of the 3′ end of the RET gene with various 5′ partner genes, referred to as PTC, leading to constitutive RET activity and increased signaling through various pathways, most notably the MAPK signaling cascade (46). Clinically, RET/PTC1 (partner gene H4) PTC have a similar clinical course of adult-onset PTC, but RET/PTC3 (partner gene ELE1) is associated with a more aggressive, solid variant of PTC.
Mouse models have consistently shown that RET/PTC rearrangements can initiate thyroid carcinogenesis. Two independent groups have generated thyroid-specific RET/PTC1 transgenic mice (25,26) (Table 1). Santoro et al. generated mice expressing RET/PTC1 under the control of the rat Tg promoter, but only about half developed PTC over a long latency period and had no distant metastasis (26). Jhiang et al. utilized the bovine Tg promoter and generated two groups of RET/PTC1 mice, low and high copy numbers of the transgene, with distinct tumor behavior (25,47). All of these RET/PTC1 mice developed PTC; the low copy number mice had a longer latency time until development of PTC (up to 6 months), whereas the high copy number group had PTC as soon as several days following birth (47). The high copy line had congenital hypothyroidism and it is interesting to note that thyroid hormone replacement could delay tumor development and progression, suggesting the importance of TSH stimulation in this model. Metastasis was also absent in both mouse lines. In mice, RET/PTC1 cancers requires additional mutations, such as knockout of the tumor-suppressor p53, to result in metastasis (48).
Transgenic RET/PTC3 mice develop PTC, similar to the human solid variant of PTC, and about one-third develop axillary lymph node metastasis unlike their RET/PTC1 mouse counterparts (49). On comparing the genetic profiles by microarray analysis of these murine tumors to human PTC, the similarities were most congruent when using mouse PTC tumors at a young age (i.e., 2 months) (50). In particular, mouse and human PTCs had similar patterns of overexpression of genes involved in cell cycle, DNA repair, and genes associated with the immune response (50). As proof-of-concept that these mice are a model for human PTC, both also had similar patterns of expression for proteins involved in the MAPK pathway. As these RET/PTC mutations are germline in mice versus acquired in humans, PTC development and tumor progression may be more advanced in mice and therefore younger specimens may correlate better with human tumors. Nonetheless, RET/PTC mouse models have expanded our ability to understand the initiators of PTC.
NTRK1
A second chromosomal rearrangement involving the neurotrophic tyrosine kinase receptor, type 1 (NTRK1), has been found in PTC (37). NTRK1 rearrangements (TRK) include diverse partner genes such as TPM3, TPR, and TPG known as -T1, -T2, and -T3 which result in constitutive activation of this receptor and overactivation of diverse pathways, including MAPK (51). These chromosomal rearrangements are less common in sporadic PTC than RET/PTC, but also suggest how ionizing radiation is detrimental to thyroid. The TRK-T1 transgenic mouse model was important as in vitro TRK-T1 cannot transform cells, and therefore, it was not clear as to its importance in thyroid cancer. About half of the transgenic mice that expressed TRK-T1 developed thyroid cancer, either FTC or PTC, without distant metastasis (15). When crossed with knockout mice of the cell cycle inhibitor, p27, these double-mutant mice developed thyroid cancers at an earlier age (52). These studies show that TRK-T1 is oncogenic and that loss of p27 may facilitate PTC development. Interestingly, TRK-T1 mice have been used to evaluate whether sonographic features used for human thyroid nodules can predict malignancy in mice. Similar to humans, mouse thyroid nodules with at least two of the following features were classified as malignant: hypoechoic, microcalcifications, poor margins, irregular shape, and extension outside the thyroid gland (22).
BRAFV600E and the MAPK pathway
The evolutionarily conserved MAPK signaling pathway is clearly important in PTC as evidenced by the RET/PTC mouse models. The MAPK pathway allows external signals to be transmitted to the cell through a signaling cascade leading to RAS activation, followed by RAF recruitment and activation which then activates the MAPK kinases (23) that phosphorylate and activate MAPK proteins (e.g., ERK) (Fig. 2). Dysregulation of MAPK signaling is common to many tumors, and mutations of BRAF are only second to RAS in terms of prevalence in tumors (53). The activating point mutation T1799A leads to a valine to glutamate substitution at position 600 of BRAF (BRAFV600E ) and is commonly found in PTC (54,55). BRAFV600E is also important for prognosis as it is associated with the aggressive tall-cell variant of PTC and/or extrathyroidal extension and dedifferentiation (55).
To understand how BRAFV600E affects PTC behavior, Knauf et al. created transgenic BRAFV600E mice under control of the bovine Tg promoter (Tg-BRAF) (27). Two distinct lines, Tg-BRAF2 and Tg-BRAF3, were studied, which differed in their level of BRAF expression. Interestingly, both lines had TSH elevation compared with wild-type mice, but the total thyroid hormone levels were the same. Goiters and PTC with similar histopathologic features of human BRAF-positive PTC occurred (Fig. 3). Higher expression of BRAF was associated with more aggressive PTC, including local invasion, but none of the mice developed distant metastasis. One clinical question that may be addressed by mouse models is how much TSH contributes to limiting aggressive thyroid cancer progression, such as tall cell. One issue with mouse models of thyroid cancer is that the Tg promoter is also regulated by TSH. Therefore, in this model and others, the higher TSH levels also led to increased oncogene expression which is not similar to human PTC (27). Recent work by Fagin and coworkers has created a mouse with doxycycline-inducible thyroid expression of BRAFV600E (Tg-rTta/TetO-BRAFV600E) which develops thyroid cancer in the presence of doxycyline and reversion of the malignant phenotype with removal of doxycycline and subsequent removal of BRAFV600E (56). The overall shared features between mouse and human BRAF-positive tumors should provide additional insight for PTC.
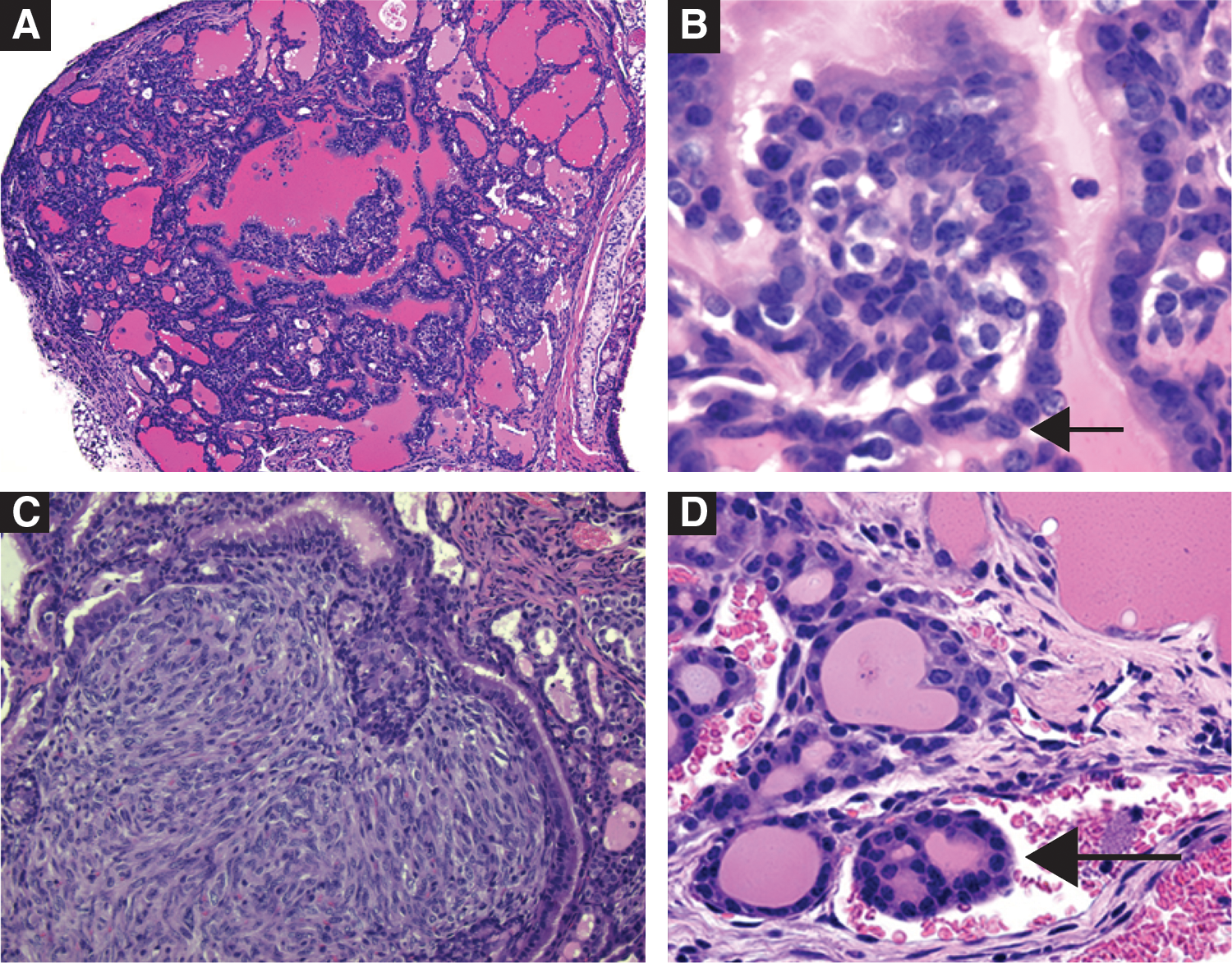
Tg-BRAF2 mice recapitulate papillary thyroid cancer (low-magnification,
FTC
FTC is a diagnostic challenge because of the inability to identify FTC on routine cytological examination by fine-needle aspiration biopsies of thyroid nodules. There has been significant progress in identifying genetic abnormalities in FTC, but none can be reliably used yet for diagnosis. Point mutations of RAS and rearrangement of the paired box 8 (PAX8)-peroxisome proliferator-activated receptor
FTC, follicular thyroid cancer; cAMP, cyclic adenosine monophosphate.
RAS
RAS is a guanine nucleotide-binding protein that is a key intermediate in signal transduction pathways. RAS mutations are found in 40–50% of conventional follicular carcinomas and in 20–40% of adenomas (67 –69). The most frequently affected hotspots are NRAS codon 61 and HRAS codon 61, although mutations of RAS involve codons 12, 13, and 61 of all three HRAS, KRAS, and NRAS genes (28,70). RAS activation depends on whether it binds to guanosine diphosphate (GDP) or guanosine triphosphate (GTP). It is inactive when it binds to GDP; when it binds to GTP, RAS initiates a downstream signaling pathway (71) (Fig. 2). The loss of the activity in the conversion of GTP to GDP by RAS mutants results in constitutive activation of signaling in this pathway (71). The presence of a RAS mutation has been found to correlate with tumor dedifferentiation and a less favorable prognosis (72). The aggressive biological properties of these tumors may be due to the effect of the mutant RAS protein on promoting chromosomal instability, which has been demonstrated in vitro (73).
Transgenic mice expressing a mutated and activated H-RasG12V controlled by the bovine Tg promoter developed PTC with concurrent lung nodules; however, these lung lesions had a papillary architecture but more likely represented primary versus metastatic tumors (74). Although different founder strains were generated, with varying levels of RAS expression, these mice were limited in that they could not pass this phenotype to their progeny. An earlier mouse model containing a mutant K-RasG12V gene under the control of rat Tg promoter showed no thyroid cancer with normal thyroid hormone levels. Following treatment of these mice with goitrogens for 6 months, only one mouse developed FTC (75) (Table 2). This suggests that RAS mutation alone is not sufficient to induce FTC but acts as a predisposing factor and that multiple genetic alterations are required for FTC (75). K-RasG12V mice could be used for the study of genetic factors that might promote the development of FTC.
Another mouse model was created by targeting a human NRAS with a mutation at codon 61 (N-RasGln61Lys ) to thyroid follicular cells (76). Comparison of thyroid histopathology between these transgenic mice and wild-type littermates up to 18 months of age indicated progressive changes from hyperplasia to adenoma and carcinoma in some transgenic mice. Among 88 mice examined, 26 (30%) developed carcinomas that were follicular or had mixed papillary–follicular features and 9 (10%) had invasive carcinomas with large poorly differentiated areas closely resembling those observed in human patients. Distant metastases were observed in the liver of three mice, lung of two mice, and the right femur of one mouse (76). Although occurrence of metastasis requires long latency, the mice are potentially useful for studies of thyroid carcinogenesis.
A concern regarding these transgenic mouse models of FTC is that the cancer phenotype resulting from overexpression of a mutated RAS gene might not reflect the activity of endogenous mutant RAS expressed at the normal physiological level. It is not clear among these models whether the differences resulted from different functions of mutant genes, different expression levels, or other factors. Accordingly, a mouse model was created to express K-RasG12D under control of the endogenous KRAS promoter (34). Similar to other models, constitutive activation of K-RasG12D did not cause any physiologic or functional thyroid alterations. As aberrant activation of the PI3K pathway has been identified in most thyroid cancers (77,78), double-mutant mice with knock-in mutated K-RasG12D gene with Pten–/– deletion targeted to the thyroid were created to examine whether mutant KRAS requires simultaneous PI3K activation to fully realize the cells' oncogenic potential. The concurrent activation of K-RasG12D and PI3K in thyroid follicular cells led to aggressive, invasive, and metastatic follicular carcinomas (34). Fifty percentage of the mice died within 7 weeks after birth, and none survived longer than 4 months. Serum TSH level was drastically reduced and T4 was increased in these mice, suggesting thyroid autonomy. All mice surviving longer than 12 weeks developed lung metastases (34). The K-RasG12D mouse model would be useful to study the interaction of different pathways necessary for the development of metastatic thyroid cancer.
TRβPV
The TRβPV knock-in mouse was initially created to model an inheritable disease called resistance to thyroid hormone (RTH) (79). The hallmark of RTH is elevated serum thyroid hormone levels associated with nonsuppressible TSH (80,81). RTH is due to the mutations of the thyroid hormone receptor β (TRβ) gene. Most RTH patients are heterozygous with a mutated TRβ allele. Although the clinical symptoms of RTH are generally mild, one patient with a homozygous mutation died at a young age with extraordinarily high levels of TSH (80,81). TRβPV mice were created through homologous recombination by targeting a PV mutation to the TRβ gene (79). The PV mutation was identified in a patient with RTH (82) who has a frameshift mutation in the C-terminal 14 amino acids of TRβ, resulting in a complete loss of T3 binding and transcriptional capacity. TRβPV mice faithfully recapitulate human RTH (79).
Remarkably, as homozygous TRβPV (TRβPV/PV ) mice age, they spontaneously develop FTC with a pathological progression similar to human FTC. As early as 2 months of age, TRβPV/PV mice exhibit papillary hyperplasia. By 4–5 months of age, signs of FTC such as capsular and vascular invasion are already apparent. Distant metastases to the lungs and heart are common in mice older than 5 months (Fig. 4). The pathological changes progress from hyperplasia (100% of all mice examined), to capsular invasion (91%), vascular invasion (74%), anaplasia (35%), and/or distant metastasis (30%). Metastasis involves mainly the lung and occasionally the endocardium, but not local lymph nodes. The small, round, dark nuclei of the developed thyroid carcinomas are characteristic of follicular carcinoma (83). The TRβPV/PV mouse model has facilitated the identification of altered signaling pathways underlying FTC. Indeed, similar to human FTC, overactivation of tumor promoters [e.g., the pituitary transforming tumor gene, cyclin D1, and β-catenin (84 –94)] and tumor-promoting signaling pathways (e.g., PI3K-AKT signaling) has been uncovered. The TRβPV/PV mouse serves as a preclinical model of FTC, having been used to test several potential therapeutic targets (85,92).
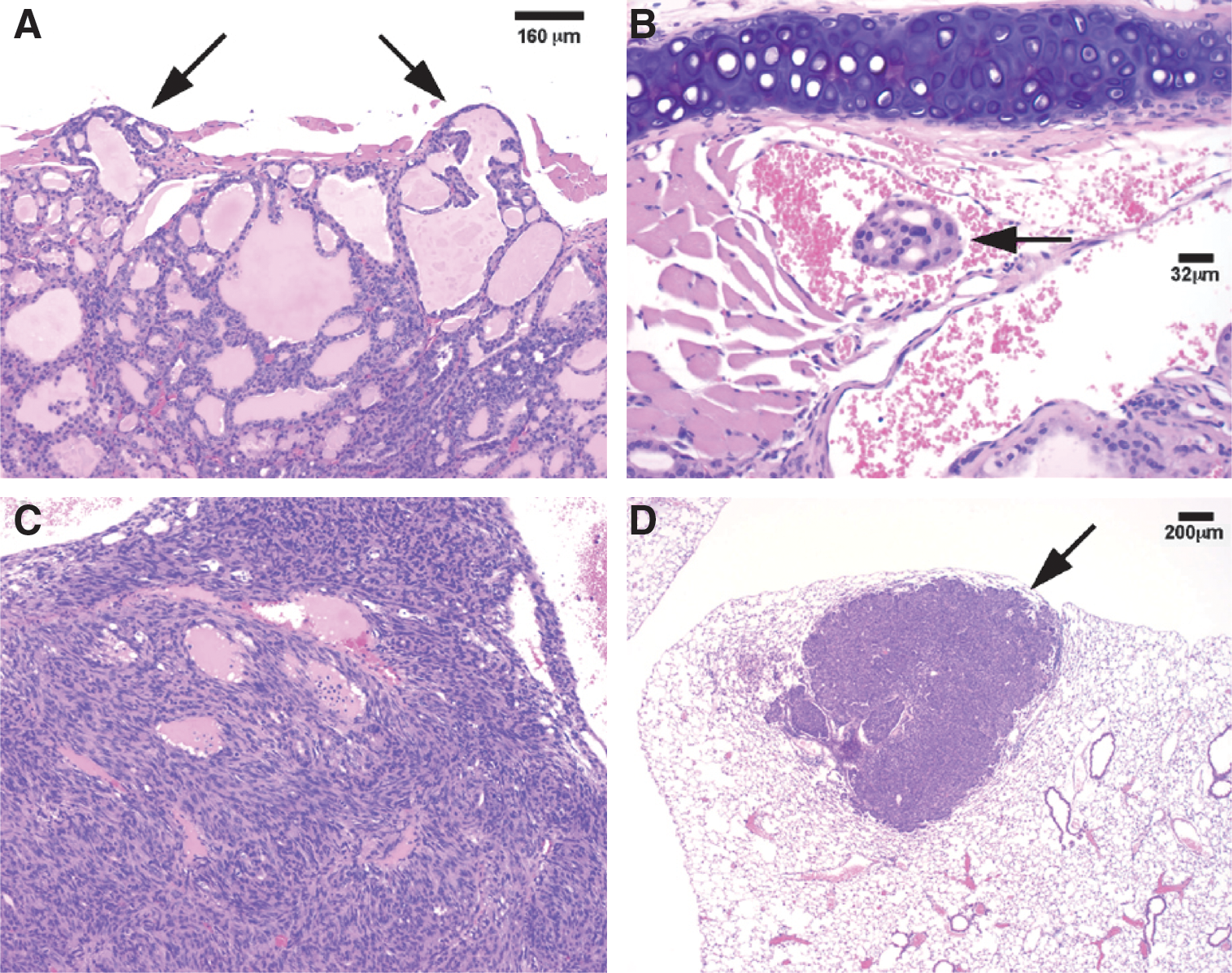
TRβPV/PV
mice recapitulate follicular thyroid cancer with capsular and vascular invasion (panels
Spontaneous development of FTC has also been observed in mice with one mutated TRβPV but without the wild-type TRβ allele (TRβPV/− mice) (95). This TRβPV/− mouse was generated by cross-breeding TRβPV/+ mice with TRβ knockout mice (95). The pathological progression in the thyroid of TRβPV/− mice was indistinguishable from that of TRβPV/PV mice. These results indicate that in the absence of a wild-type allele, the mutation of one TRβ allele is sufficient for the mutant mice to spontaneously develop FTC. These results suggest that the TRβ could function as a tumor suppressor. Importantly, these findings raise the possibility that TRβ could be tested as a novel therapeutic target in thyroid cancer.
The TRβPV/PV mouse is a unique model, as it closely resembles human FTC in pathological progression and its underlying genetic alterations (84 –94,96,97). Though it is not known whether the only reported patient with homozygous mutation of the TRβ gene (81) developed thyroid cancer, abnormal expression or somatic mutations of TR are associated with several human cancers including PTCs (98,99). The silencing of the TRβ gene by hypermethylation and the concurrent reduction of TRβ1 transcripts were demonstrated in breast cancer (100). In cotransfection experiments, both TRα1 and TRβ1 strongly repressed HRASG12V -induced transformation of NIH3T3 fibroblasts, reduced tumor volume, and inhibited tumor growth in nude mice (101). Also, the transfection of TRβ1 into hepatocarcinoma and breast cancer cells reduced tumor growth, caused partial mesenchymal-to-epithelial cell transition, and had a striking inhibitory effect on invasiveness, extravasation, and metastasis formation in mice (102). The results from the TRβPV/PV mouse model are consistent with these reports that the TRβ receptors could act as a tumor suppressor in the development of FTC.
Mutant α1B-adrenergic receptor
A mutated α1B-adrenergic receptor gene under control of the bovine Tg promoter resulted in constitutive activation of the TSH signaling pathway and was used to establish a model for FTC (103). In the first few weeks of life, the mice began to develop goiters and by 12 months some thyroids were 130 mg which is over 20 times normal. As the mice aged, thyroid nodules developed, and in a few cases, all morphological criteria of thyroid follicular cell differentiation were lost; spindle-cell foci were found without formation of identifiable follicles. The foci frequently reached malignancy criteria for human thyroid tumors with local invasion into the muscle. Lung metastases were observed at an overall rate estimated to be 20% in mice older than 12 months. The manifestation of advanced tumor features in this mouse model may be useful for studies of the invasion and metastasis of FTC mediated by TSH-activated signaling pathways.
Rap1bG12V
RAP1 is a member of the RAS family of small G proteins that transmit signals from the TSH receptor to the MAPK pathway by means of a cAMP-dependent mechanism in thyroid cells (104). RAP1 was shown to have both mitogenic and tumorigenic properties (105). In a mouse model, a thyroid-specific constitutively active Rap1b (Rap1bG12V) also allowed the inducible switch to the inhibitory form of Rap1b (Rap1bS17N) (33). Mice expressing Rap1bG12V showed no signs of hypothyroidism or hyperthyroidism. Exposure of Rap1bG12V mice to a 6-month goitrogen administration led to nodular thyroid enlargement which reversed after goitrogen removal. When expression of the Rap1b gene was switched to the inactive Rap1bS17N , the thyroid gland size decreased by 50% despite continued goitrogen treatment, because of decreased proliferation versus increased apoptosis. However, after long-term goitrogenic treatment for 12 months, some Rap1bG12V , but no wild-type mice, developed signs of FTC without metastasis. Rap1bG12V appears oncogenic in the presence of elevated TSH-mediated signaling. The Rap1bG12V mouse model might be useful in studies of multiple factors that could contribute to the development of thyroid cancer.
Conclusions
The development of these mouse models of thyroid cancer have not only confirmed the transforming ability of single-gene alterations, such as RET/PTC translocations or B-RAFV600E , in initiating thyroid cancer, but have also shown new genes responsible for FTC, such as TRβ, and the importance of understanding additive and synergistic gene effects on thyroid cancer, such as PTEN deletion and KRAS overactivation.
Clinically, the presence of distant metastasis is the most important factor in determining a patient's prognosis. It is interesting to note that the models of PTC do not develop distant metastasis unless another mutation is introduced, whereas in FTC, three different mouse models of thyroid cancer with lung metastasis exist: TRβPV/PV , a mouse model with simultaneous activation of RAS/PI3K pathway, and a mouse model of the mutant α1B-adrenergic receptor (34,83,103). This may parallel human thyroid cancer, as FTC is more likely to have distant metastasis than PTC, or suggest a common pathway required for thyroid cancer metastasis. All three FTC models with metastasis share the features of an enlarged thyroid and an elevated TSH-mediated signaling pathway. In mouse models with RAS mutations, elevated TSH levels often promote the development of FTC. However, the precise role of TSH in thyroid carcinogenesis and whether it plays a more critical role in FTC tumorigenesis versus PTC remain to be fully elucidated.
Mouse models of thyroid cancer are important for developing therapeutic interventions. The roles of several signaling pathways, such as RAS-MAPK, and PI3K-AKT, in thyroid cancer are better understood by using transgenic mouse models. With the generation of mouse models of thyroid cancer, pharmaceutical agents may be developed and evaluated in vivo and on primary cell cultures from these mouse tumors prior to use in clinical trials. One of the limitations of many clinical trials for thyroid cancer is the inability to compare tumors pre- and posttreatment, to understand how targeted agents such as TKIs alter tumor phenotype. Coupled with radiologic imaging, mouse thyroid cancers could potentially be followed for response to a targeted agent, and tissue could be obtained before and after treatment for analysis. These preclinical models of thyroid cancer have opened many new and exciting research avenues which may have direct benefit to thyroid cancer patients in the near future.
Footnotes
Acknowledgments
This work was supported in part by a grant from the Thyroid, Head and Neck Cancer Foundation/American Thyroid Association (to C.S.K.) and the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (to X.Z.). The authors regret any reference omissions due to length restrictions.
Disclosure Statement
No competing financial interests exist.