
Abstract
Background:
The prevalence of congenital hypothyroidism (CH) increased during the period 1994–2002 in Japan. The reasons for these recently described increases in the prevalence of CH remain unclear. Moreover, the proportion of patients with different etiologies CH in the more recently diagnosed patients has not been established. In this study, we determined the etiologies of CH that were detected by neonatal screening in Niigata Prefecture, Japan.
Methods:
A total of 100 patients having a diagnosis of CH (41 men and 59 women, aged 5–19 years old) were evaluated. To determine the etiology of CH, the patients underwent a 123I thyroidal radioiodine uptake test, a scintigram, a saliva to plasma radioiodine ratio analysis, a perchlorate discharge test, thyroid ultrasonography, measurements of thyroidal function and thyroglobulin, and a thyrotropin (TSH)-releasing hormone tolerance test.
Results:
Patients with overt CH (n = 34, elevated TSH levels with low free thyroxine levels) made up 34% of the total group, 56% of the patients had subclinical CH (n = 56, elevated TSH levels with normal free thyroxine levels), and 10% had normal thyroid function. These were patients who were considered to have transient hypothyroidism or hyperthyrotropinemia. Thyroid dysgenesis was the diagnosis in 73% of patients with overt CH, and the most of these had ectopic thyroid tissue. In contrast, thyroid dysgenesis was the diagnosis in only 36% of the patients with subclinical CH.
Conclusions:
Only 50% of our patients with CH detected by neonatal screening had thyroid dysgenesis. With an increase in the percentage of patients with subclinical hypothyroidism, the prevalence of thyroid dyshormogenesis has increased. Studies of the frequency and etiology of CH should consider overt and subclinical CH separately.
Introduction
Neonatal screening for CH was initiated in Japan in 1979. Since then, almost all newborns have been tested for CH in Japan. In 1989, a highly sensitive method for detecting CH was introduced, and by 1992, an enzyme-linked immunosorbent assay had completely replaced older methods such as radioimmunoassay and enzyme immunoassay. During the period 1994–2002, the incidence and prevalence for CH in Japan had an upward trend despite the fact that the criteria for the diagnosis of CH, and screening methods including the TSH assay, did not change (4). A report published in 2004 suggested prenatal iodine exposure as a possible reason for this trend (5) but this, or other reasons for the trend, have not been established. It is also not known what etiologies accounted for CH in patients born from about 1994 to recent years.
In the present study, we sought to determine the etiology of CH in patients born between 1989 and 2005 in Japan, using the Niigata prefecture of Japan as our sampling area.
Methods
Between April 1989 and March 2006, 437,867 newborns were screened for CH in Niigata prefecture. Blood samples were collected on filter paper within the first 4–7 postnatal days. TSH was determined in single 0.3 cm disks punched from the filter paper blood. TSH was measured in the filter paper sample using an enzyme-linked immunosorbent assay (TSH: Enzaplate N-TSH; Bayer Co.). Detection limits were 0.5 mU/L for TSH. All CH screening tests are centralized in the Niigata Health Laboratory center.
In the Niigata prefecture, a blood TSH cutoff level of 8 mU/L was established for the initial neonatal screen when the program was started in 1982. When the initial blood TSH was between 8 and 30 mU/L, a second specimen was reevaluated at Niigata Health Laboratory center. If that specimen was also greater than 8 mU/L, the policy was that a confirmatory test be run within 30 days at the patient's medical institution. If the initial blood TSH was in excess of 30 mU/L, the policy was that a confirmatory test be run within 14 days at the patient's medical institution. The tests that were run at the patient's medical institution in addition to repeating the TSH was a thyroxine (T4) or free T4 (FT4) and free triiodothyronine (FT3). In addition, thyroid morphology was evaluated by ultrasonography in almost all patients. Neonates with persistent serum TSH values over than 10 mU/L and normal or low FT4 were considered to have CH, and treatment was generally started. Neonates with serum TSH values less than 10 mU/L were considered to have normal thyroid function.
Among the 437,867 newborns who were screened, 367 were considered positive for CH and were referred to pediatric endocrinologists. Of these, 333 (91%) were seem at our institution, 240 of whom were started on oral levothyroxine therapy for CH. Of the remaining patients, 86 had normal thyroid function and 7 had a chromosomal abnormality. Most patients with CH were reevaluated at 2 years of age to see whether they continued to need levothyroxine. If their serum TSH was less than 5 mU/L while off thyroid hormone replacement for 4 weeks, they were considered to have had transient CH or hyperthyrotropinemia. Of the 240 patients who had been started on levothyroxine during infancy, 12 were lost to followup, 57 were diagnosed as having transient CH or transient hyperthyrotropinemia, and in 171 the diagnosis of CH was confirmed. These latter 171 patients were the ones eligible for our study of the etiology of CH. Among these 171 patients with CH, 100 patients (58% of the eligible patients, 41 boys and 59 girls) actually participated in the study. Most of the 71 patients who did not participate could not take a capsule orally or remain in the supine position for the duration of a radioiodine uptake (RAIU) test. The 100 patients who participated were born between 1989 and 2005 and ranged in age from 5 to 19 (5–9 years old; n = 53, 10–14 years old; n = 33, 15–19 years old; n = 14) at the time of the study of the current status of their diagnosis of CH, which was based on earlier screening guidelines for CH (6). Studies were performed between April 2004 and March 2008.
Protocol for determination of etiology of CH
Levothyroxine treatment was stopped at the beginning of the study and T3 was started in three daily doses, the total of which was 25% by weight of the levothyroxine dose the patient had been on. This was given for 3 weeks. After this, there was a 7-day period during which no thyroid hormone was administered and then a 123I thyroidal RAIU, saliva to plasma radioiodine ratio, perchlorate discharge test (if the RAIU was 20% or more), scintigram, thyroid ultrasonography, and measurements of serum TSH, FT4, FT3, T3, T4, and thyroglobulin were performed. A thyrotropin-releasing hormone (TRH) stimulation test (7 μg/kg intravenous Protirelin®, HIRTONIN; Takeda Pharmaceuticals Ltd.) was also performed. Blood samples were drawn at 0, 30, 60, 90, and 120 minutes for TSH, and at 0 and 120 minutes for T3, and FT3. The T3 and FT3 increments after the TRH test (ΔT3, ΔFT3) were compared with normal values (ΔT3>25 ng/dL, ΔFT3>0.6 pg/mL) (7,8). FT3, FT4, and TSH were determined by chemiluminescent enzyme immunoassays (E-test Tosoh II kit; Tosoh Corporation). Normal ranges in our laboratory are FT3 = 2.1 to 4.6 pg/mL, FT4 = 0.9 to 1.6 ng/dL, TSH = 0.6 to 4.2 mU/L, and peak TSH levels during the TRH test are <30 mU/L. Thyroid ultrasonography was performed with LOGIQ S6 (GE health care) equipment with high-frequency probes (10 MHz). The maximum thickness and maximum width of the bilateral lobes were measured by transverse scans. The sum of thickness and the sum of width were used to estimate thyroid gland size, as a function of height (9). Thyroid hypoplasia and hyperplasia were defined as values lower than −1.0 SD and higher than +1 SD from the normal mean, respectively.
Based on thyroid ultrasonography and scintigraphy, patients with CH were classified as having thyroid agenesis, ectopy, hemiagenesis, hypoplasia, a normal-size thyroid, or hyperplasia according to thyroid morphology. Patients with thyroid hyperplasia were classified as having dyshormogenesis and were sub-classified as follows. If their saliva to plasma radioiodine ratio was 10 or less, they were considered to have an iodine concentration defect. If the perchlorate discharge test was more than 20%, they were considered to have a complete iodide organification defect, and if it was 10%–20%, they were considered to have a partial organification defect. If the serum thyroglobulin concentration was low (thyroglobulin <10 ng/mL), they were considered to have a thyroglobulin synthesis defect. Patients with thyroid hyperplasia who did not have any of these conditions were considered to have a post iodide organification defect in thyroid hormone synthesis. Among patients who had normal thyroid glands, patients who had a less than normal T3 or FT3 response to TRH were considered to have resistance to TSH (RTSH) and were classified as having dyshormonogenesis. All of the patients in this classification had a TSH response to TRH. Those in the normal thyroid size group who did not have RTSH were considered to be “idiopathic.” Thyroid agenesis, ectopy, hemiagenesis, and hypoplasia were categorized as thyroid dysgenesis.
All 100 patients who enrolled in the study had, as previously noted, a previous history and tests that supported the diagnosis of CH. Nonetheless, not all patients who were evaluated by the study protocol had overt or even subclinical hypothyroidism (see Results). On the basis of the study protocol results, patients were divided into three groups based on their serum TSH and thyroid hormone levels after they had been off thyroid hormone for one week. Patients who had elevated basal TSH levels or whose serum TSH increased to 30 mU/L or higher during the during TRH test but who had normal serum FT4 concentrations were assigned to the subclinical hypothyroid (SCH) group. Patients who had elevated serum TSH concentrations and low serum FT4 concentrations were assigned to the overt hypothyroid (OH) group. Patients who had normal serum TSH and FT4 concentrations were assigned to the transient hypothyroid (TH) group.
The study was approved by the Institutional Review Board Committee at Niigata University School of Medicine, and informed consent was given by all participants in this study or by their parents or guardians on their behalf.
Results
In all patients, peak serum TSH levels were found 30 minutes after TRH administration and declined thereafter. The etiologies of CH in the study patients are shown in Table 1 and Figure 1. The OH group made up 34% of the patients with CH, the SCH group made up comprised 56% of the patients with CH, and the TH group comprised 10% of the patients with CH. The distribution of FT4 and TSH values in the three groups are shown in Figure 2. Thyroid dysgenesis accounted for 73.4% of the OH group, and almost all had thyroid ectopy. In contrast, thyroid dysgenesis accounted for 35.7% of the SCH group, and half of the SCH group had normal thyroid position and size without typical thyroid dyshormogenesis and were classified as idiopathic.
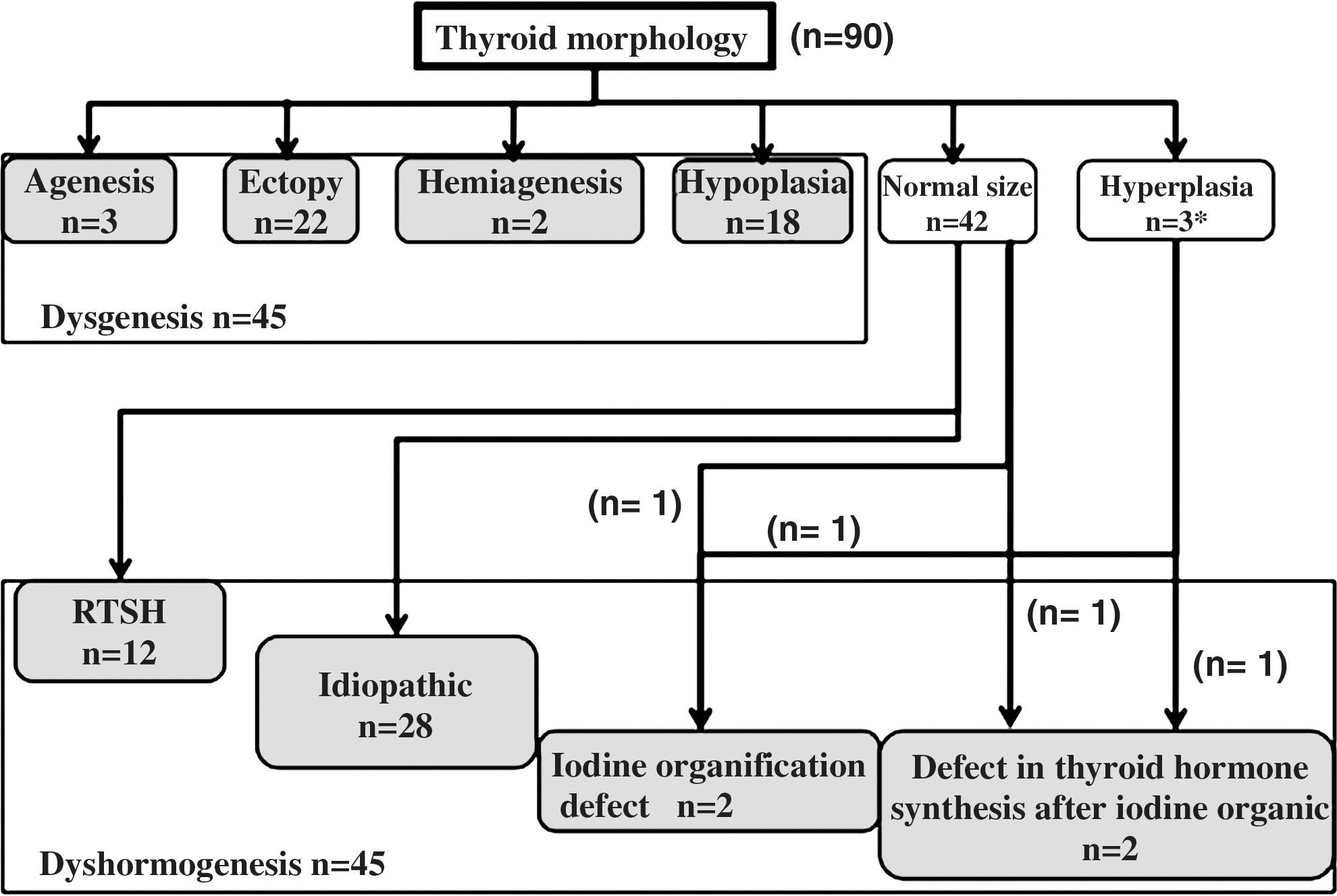
Classification of the etiology of congenital hypothyroidism. n shows the number of patients with congenital hypothyroidism. Among normal sizes, cases of less than normal value ΔT3 and ΔFT3 on thyrotropin-releasing hormone tests are classified as clinical resistance to thyrotropin (RTSH). Normal sizes without RTSH or typical dyshormogenesis were classified as “idiopathic.” *One patient had coexistence of Hashimoto disease.
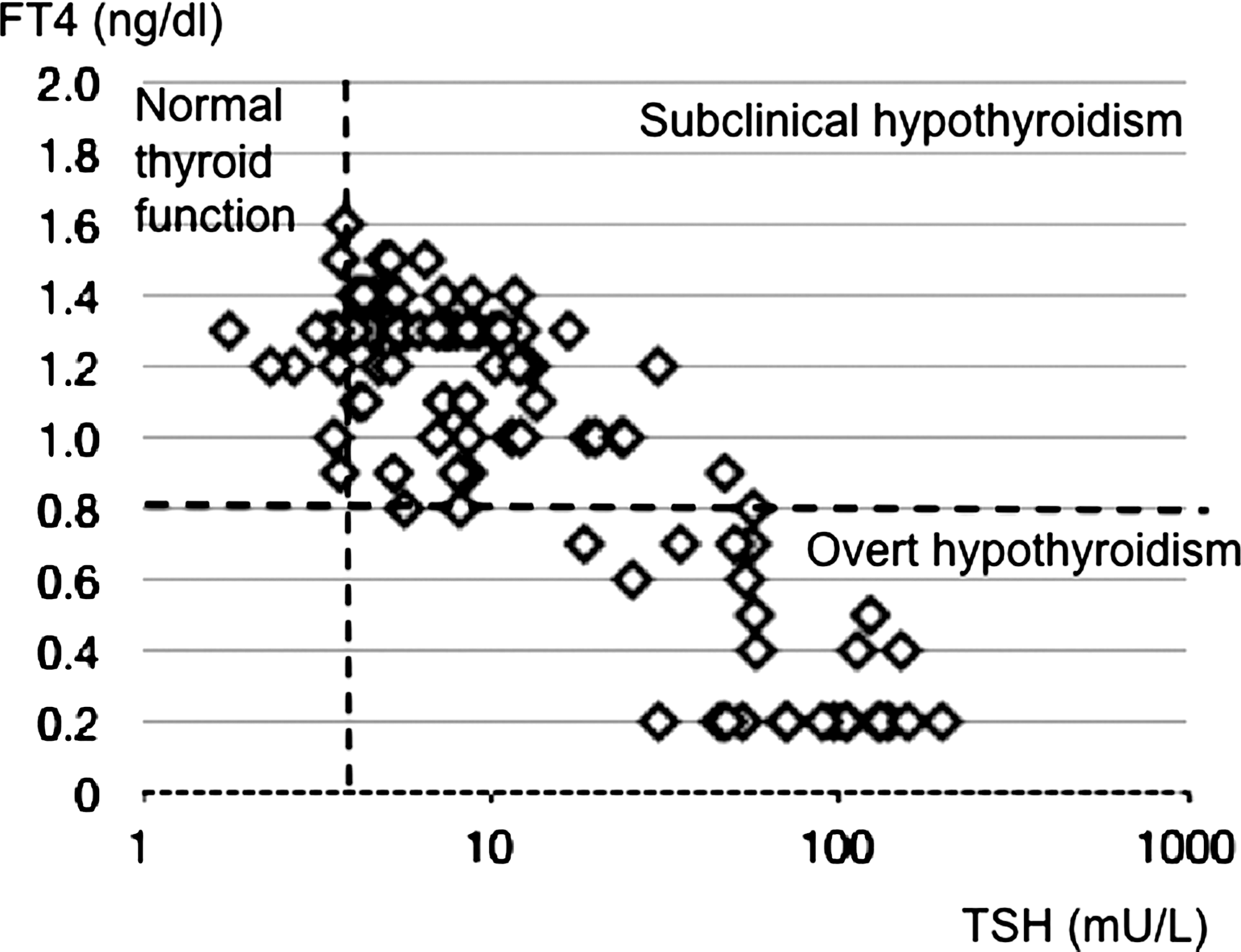
The distribution of free thyroxine (FT4) and TSH values in the three groups after the withdrawal of replacement in this study.
Coexistence of Hashimoto disease.
Among patients with SCH, almost all patients except one had a normally located thyroid gland. Clinical RTSH was detected in 12 patients and was found in both patients with OH and those with SCH.
Discussion
This was the study of the etiologies of CH as detected by neonatal screening in Japan. In 10 of the 100 study patients who carried a diagnosis of CH, this diagnosis could not be confirmed. Of the remaining 90 patients, 45 (50%) had thyroid dysgenesis, and 45 (50%) had thyroid dyshormogenesis patients. This contrasts with earlier findings that the majority of CH cases (about 75%–85%), had some form of thyroid dysgenesis, including agenesis, ectopy, hemiagenesis, or hypoplasia (1,2) with the remaining patients having thyroid dyshormogenesis. These epidemiological and clinical classifications, however, were based on screening programs with TSH cutoff values of 20–40 mU/L or first performing T4 screening with backup TSH measurements in the dried blood spot. In contrast, our initial screen was based on a TSH cutoff value of 8 mU/L, which likely detected more of the milder forms of CH. These were mostly subclinical hypothyroidism. We classified patients in whom the diagnosis of CH was confirmed by the study as SCH or OH. About 73% of the OH patients with OH had thyroid dysgenesis in agreement with earlier reports (1,2); approximately 90% of those were ectopic. In contrast, only 36% of the SCH group had thyroid dysgenesis. In future studies, we recommend that OH and SCH forms of CH be considered separately, but followup studies of patients initially screened using relatively high blood spot TSH concentrations may be relatively underrepresented by patients with SCH.
RTSH was detected in 12 patients who were in both the SCH and OH groups. RTSH is a heterogeneous condition where there is a variable degree of insensitivity to biologically active TSH. In this study, we defined clinical RTSH based on the incremental increase in serum FT3 after TRH administration. A similar approach was taken by Balavoine et al. (10). Unfortunately, there is very little definitive reference data for TRH stimulation tests, and TRH is no longer marketed in many regions. RTSH is classically caused by loss-of-function mutations of the TSH receptor gene (TSHR), but some patients exhibit a RTSH-like phenotype in the apparent absence of TSHR mutations. Some have mutations of PAX8, GNAS1, or TITF1 (11). Patients with mutations of TSHR have a broad clinical spectrum, depending on the degree of TSH unresponsiveness, ranging from subclinical to overt hypothyroidism (12). Some patients have hypoplastic thyroid glands; in others, thyroid size is normal. A more accurate etiologic study would require an active genetic analysis in addition to a conventional etiology of CH.
Almost half of our patients in the SCH group were classified as “idiopathic” on the basis of normal baseline FT4, elevated TSH levels, a normal T3 response to TRH, and normal thyroid images. Many pediatric endocrinologists do not consider patients of this type to necessarily have permanent hypothyroidism (13). This category includes various conditions such as mild dyshormogenesis, a thyroid gland in the lower limits of normal size, and mild resistance to TSH,. These are classified as dyshormogenesis according to conventional classification, as they all have a normal thyroid gland position. Further studies to define more accurately the underlying pathophysiology in these patients and whether they are longstanding are needed.
In summary, in our study of Japanese children and teenagers who carried a diagnosis of CH that was made in conjunction with a screening program with a relatively low initial TSH cutoff value, the diagnosis of CH could not be confirmed in 10% of the patients. In the remaining 90%, only 50% of these had thyroid dysgenesis and there was a relatively high proportion of those who would be judged to have subclinical hypothyroidism, at least using criteria developed in adults. Further followup studies of CH should consider both subclinical and overt hypothyroidism. In addition, the screening strategies of the various neonatal programs should be scrutinized regarding the possibility that some employ TSH cutoff values that are not sufficiently sensitive.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.