
Abstract
Background:
Resistance to thyrotropin (TSH) causes congenital hypothyroidism (CH). TSH receptor (TSHR) and adenylate cyclase-stimulating G alpha protein subunit (GNAS) loss-of-function mutations cause TSH resistance. We describe a family with TSH resistance and CH bearing a combination of inactivating mutations in TSHR and GNAS genes. We describe studies to determine the molecular mechanisms involved in TSH resistance in this family.
Methods:
DNA sequencing to identify TSHR and GNAS gene mutations was performed. In vitro effects of the mutations on cAMP production and TSH binding were investigated in COS7 cells. cAMP production was evaluated by transfecting a cAMP response element (CRE)-luciferase reporter with pSVL-TSHR and pSVK3-GNAS vectors. For binding studies, cells transfected with pSVL-TSHR vectors were incubated with iodine-125 bovine TSH (125IbTSH).
Results:
Family members with and without CH were heterozygous for the TSHR mutant p.E34K or the GNAS mutant c.750_751insA (=GNASMut). The propositus had CH and he was heterozygous for TSHR p.E34K; his mother, also heterozygous for TSHR p.E34K, did not have CH. The euthyroid propositus' wife was heterozygous for GNASMut. The propositus' two daughters had CH, one was heterozygous for GNASMut and the other a compound heterezygous for TSHR p.E34K and GNASMut. Albright's hereditary osteodystrophy phenotype was present in those with GNASMut mutation but only the daughters had pseudohypoparathyroidism type 1a. Cells transfected with TSHRE34K had lower TSH affinity and less CRE-luciferase response than cells transfected with TSHR wild-type (WT). Cells transfected with GNASMut did not stimulate CRE-luciferase activity, but when cells were transfected with GNASMut plus GNASWT, a similar response to GNASWT alone was observed. The combination of TSHRWT and GNASWT showed higher CRE-luciferase response than TSHRWT and TSHRE34K with either GNASWT or GNASWT plus GNASMut.
Conclusions:
CH was caused by loss-of-function mutations in TSHR and/or GNAS. The absence of CH in the propositus' mother argues against a role for TSHR p.E34K being the only cause of CH. The minimal thyroidal phenotypic differences between the sisters with pseudohypoparathyroidism type 1a and TSH resistance, both heterozygous for GNAS c.750_751insA but only one bearing the TSHR p.E34K mutant, suggest that the main cause for CH was preferential expression of the mutated maternal GNAS allele in the thyroid gland.
Introduction
The TSHR gene maps to the long arm of chromosome 14 (14q31), has 10 exons, and encodes a 764-amino-acid long G-protein-coupled glycoprotein that controls growth and function of thyroid cells via stimulation of adenylyl cyclase (AC) and phospholipase C (PLC) (6). TSHR has an extracellular amino-terminal domain of 418 amino acids, codified by exons 1–9, that is responsible for TSH binding; the seven-transmembrane domain and the carboxyterminal intracellular domain are codified by exon 10 and are responsible for TSHR interactions with G proteins (6). TSHR LOF mutations are distributed throughout the receptor although some mutations have been reported with higher frequencies (2). The phenotype of patients with TSHR mutations depends on the number of mutated alleles, with homozygous and compound heterozygous patients having a more severe phenotype than heterozygous patients. Phenotype is also affected by a specific functional TSHR mutations that can impair TSH binding, decrease TSHR capacity to activate G-proteins, and/or reduce its expression at the cell surface (2). The presence of different phenotypes between subjects with similar mutations, and even between individuals from the same family, suggests the involvement of additional unknown factors (2).
The G alpha protein subunit (GNAS) complex locus maps to the long arm of chromosome 20 (20q13.2-20q13.3). This locus has highly complex imprinted expression with maternally, paternally, and biallelically expressed transcripts that derive from the use of four alternative promoters and also the presence of methylated regions in their 5′ exons (7). While Gsα is the most characterized protein derived from GNAS, there are at least four more GNAS products: neuroendocrine secretory protein 55, the extra-large variant of Gsα, the A/B transcript, and the GNAS antisense transcript (8,9). Gsα is encoded by 13 exons that use alternative splicing to generate five different Gsα transcripts. There are long and short Gsα variants, based on alternative splicing of exon 3 and, since there are two different splice sites at the start of exon 4, each of these Gsα variants either includes or excludes a CAG triplette (9). The fifth transcript is Gsα-N1, a truncated variant in the C-terminus (9). The thyroid gland preferentially expresses the maternal GNAS allele (5), which explains how patients with pseudohypoparathyroidism type 1a (PHP1a), a condition caused by maternally inherited GNAS LOF mutations, presents with TSH resistance (9).
In the current report, we describe a family with several members presenting with TSH resistance associated with congenital hypothyroidism (CH) caused by a unique combination of inactivating mutations in TSHR and GNAS genes. In vitro functional studies performed to investigate the effects of these mutations on TSHR activity are also discussed.
Materials and Methods
Subjects
The family pedegree is shown in Figure 1. The propositus, a 38-year-old man, was found to have CH without goiter at 18 months of age, presenting with obesity, macroglosia, severe constipation, and listlessness (Fig. 1, subject II-3). The patient was treated with thyroid hormone replacement and reached all his developmental milestones at the expected ages. In 1991, to perform a thyroid scan, the patient's thyroid-hormone replacement treatment was stopped, resulting in the following serum levels: TSH 26.7 mIU/L (normal: 0.1–4.5), total thyroxine (T4) 1.1 μg/dL (normal: 4.5–12), free T4 (FT4) 0.18 ng/dL (normal 0.7–1.9), and total triiodothyronine (T3) 64 ng/dL (normal:60–185); the scan showed the thyroid gland to be in its expected location with a normal technetium-99m (Tc99m) distribution. The propositus's mother had hypothyroidism secondary to radioiodine treatment for Graves' disease and no serum TSH data were available before the diagnosis of hyperthyroidism. The propositus's wife (Fig. 1, subject II-4) was a 34-year-old euthyroid woman of short stature (153 cm) who was found to have heterotopic ossification in the forearms affecting the medial nerves and requiring surgical resection; she had not been found to have PHP1a, and her serum calcium was 9.1 mg/dL (normal: 8.8–10.2), phosphorus 4.2 mg/dL (normal:2.3–4.5), and parathyroid hormone (PTHi) 41 pg/mL (normal ≤ 80). The propositus's oldest daughter (Fig. 1, subject III-1), born in September 2002, was found on neonatal screening to have a serum TSH of 65.6 mIU/L (normal: 0.3–5.5), FT4 1.34 ng/dL (normal 0.85–1.8), and free T3 (FT3) 3.03 pg/mL (normal: 1.63–3.78); a Tc99m thyroid scan showed the gland to be in the expected location but with decreased uptake of Tc99m. The patient was given thyroid hormone replacement, but by the age of 5 years, obesity, coarse facial features, a wide nose, and small diffuse points of heterotopic ossifications in the face were noticed; serum calcium was 7.6 mg/dL and PTHi 839 pg/mL; the child was therefore found to have PHP1a and treated with oral calcium and vitamin D. By 7 years her height was 122.5 cm, close to the 50th percentile. The second daughter of the propositus (Fig. 1, subject III-2) was born in November 2004, presenting at birth with a temporomandibular joint ankylosis and severe restriction in opening the mouth that required surgical reconstruction; neonatal screening showed a serum TSH level of 44.85 mIU/L, FT4 1.03 ng/dL, and an FT3 2.36 pg/mL; a Tc99m thyroid scan showed the gland in the expected location and with normal morphology, but decreased uptake of Tc99m. The baby was treated with thyroid hormone replacement and by the age of 3 years had a phenotype similar to that of her sister, except with more severe heterotopic facial ossification, including plates in the cheek, chin, and eyelids. Her serum PTHi was 288 pg/mL, calcium 7.6 mg/dL, and phosphorus 5.3 mg/dL, indicating treatment with oral calcium and vitamin D; at age 5 years 3 months her height was 104.9 cm (3rd to 10th percentile).

Family pedigree and genotypes. The propositus (subject II-3), a 38-year-old man with congenital hypothyroidism
Genetic studies
Genomic DNA was extracted from blood cells (RealPure Extraction DNA kit; Durviz) of the propositus and 11 members of his family. TSHR, paired box 8 (PAX8), and GNAS coding exons and adjacent intron fragments of 50–100 bp were amplified by polymerase chain reaction (PCR) (oligonucleotide sequences and PCR conditions available upon request) and sequenced in an ABI PRISM 3100 (Applied Biosystems). Informed consent was obtained from all family members, according to the Ethics Committee of the Clinical Hospital of the University of Santiago de Compostela.
Construction of TSHR and GNAS plasmid vectors
A TSHR mutant E34K vector, pSVL-TSHR-E34K, was created by site-directed mutagenesis (QuickChange II site-directed mutagenesis kit; Stratagene) using a pSVL-TSHR-wild-type (WT) vector (a gift from S Refetoff, Thyroid Study Unit, University of Chicago) and the oligonucleotide 5′-gcgagtgccatcagaaggaggacttcagagtc-3′ as previously reported (10).
The pSVK3-GNAS-WT vector was constructed as previously described (10); this vector contains the GNAS transcript variant 3 (NCBI Reference Sequence: NM_080426.2) encoding GNASS, a short form of Gsα that lacks exon 3 and has an extra serine residue due to an alternative splice acceptor site for exon 4 of the GNAS gene. A vector with an A insertion at triplet 250 of GNASS, henceforth referred to as the pSVK3-GNAS-Mut, was constructed as follows: total RNA was extracted from blood cells of the propositus's wife using TRIzol (Invitrogen) as per the manufacturer's instructions and reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Invitrogen), 1 μg total RNA, 5 μM random hexamers, and 2 mM deoxynucleotides (Ecogen) in a 20 μL reaction volume. cDNA fragments spanning exons 1–13 were PCR-amplified using as a forward oligonucleotide primer 5′-
Cell culture, transfection, and dose–response studies
The effects of the TSHR and GNAS mutants on cellular cAMP production were investigated by cotransfection studies using a cAMP response element (CRE)-luciferase reporter plasmid (a gift from Dr Peter Syapin, Pharmacology Department, TexasTech University). COS-7 cells (simian virus 40-transformed African green monkey kidney fibroblast) were grown at 100% confluence in 100 × 20 mm polystyrene tissue culture dishes (Falcon) in Dulbecco's modified Eagle's medium (Sigma-Aldrich) containing 10% fetal bovine serum (Sigma-Aldrich) and 50 mg/L gentamicin (Sigma-Aldrich), at 37C in 100% humidity and 10% CO2. For transient transfections, cells were transferred to 24-well culture plates (50,000 cells/well; Sarstedt) and grown in 500 μL of the same medium until they reached 70%–80% confluence. On the day of transfection, the medium was removed, cells were washed twice with phosphate buffered saline (PBS), and to each well 500 μL fresh medium was added, followed minutes later by Lipofectamine Reagent (Invitrogen; 1.25 μL/well) and CRE-luciferase reporter plasmid (250 ng/well), together with pSVL-TSHR vectors (Fig. 2) and additionally in some experiments pSVK3-GNAS vectors (Figs. 4 and 5). The total amount of expression vectors was 250 ng/well and in combination equal proportions of each vector were maintained to make up to the total of 250 ng/well. After 48 hours, recombinant human (rh)TSH (Thyrogen, a gift from Genzyme Corporation) was added at the following doses: 0, 0.05, 0.1, 0.5, 1, 5, 10, and 100 IU/L. The cells were incubated for a further 8 hours. Response to rhTSH was determined using a Luciferase Assay System (Promega), and luciferase activity measured with a Junior LB 9509 luminometer (Berthold). Each plasmid was transfected in triplicate and each experiment repeated at least twice.
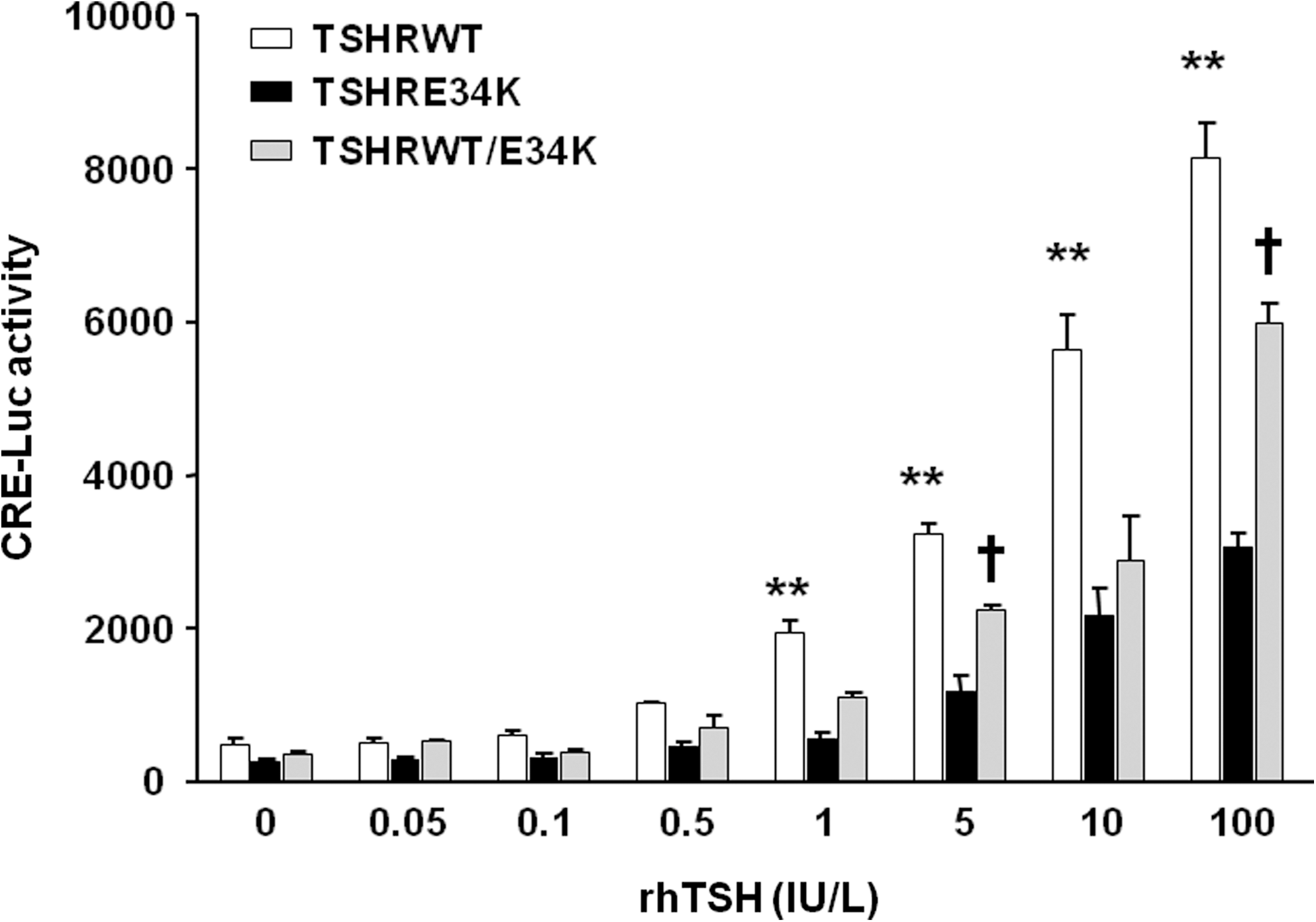
Dose–response curves for CRE-luc activity induced by different doses of rhTSH in COS-7 cells transiently expressing TSHR wild-type and p.E34K mutant receptor. Data are plotted as average ± SE, for two experiments; each point was run in triplicate. Data were analyzed using GraphPad Prism software. RLU, relative luciferase unit; rhTSH, recombinant human TSH.
Binding assays
Bovine TSH (bTSH; Sigma-Aldrich) radio-iodination was carried out by the chloramine-T method as previously reported (11). Binding studies were carried out in COS-7 cells transfected with 500 ng/well pSVL-TSHR-WT or pSVL-TSHR-E34K vectors. About 48 hours after transfection, cells were washed twice with modified Hank's buffer (5.36 mM KCl; 0.44 mM KH2PO4; 0.41 mM MgSO4; 0.33 mM Na2HPO4; 5.55 mM glucose) supplemented with 220 mM sucrose. Cells were incubated at room temperature in this buffer (supplemented with 2.5% low-fat milk) containing about 100,000 counts/mL 125I-bTSH and increasing concentrations of unlabeled bTSH (0, 0.01, 0.03, 0.1, 0.3, 1, 3, 10, 30, and 100 mU/mL). Four hours later, the cells were washed twice in the same modified Hank's buffer and solubilized by treatment with 1 N NaOH. Bound radioactivity was determined in an automatic gamma-counter (1470 Wizard Wallac PerkinElmer).
Nucleotide and amino acid nomenclatures
The nucleotide positions in human TSHR and GNAS mRNA were designated according to reference sequences (GenBank accession number NM_000369 for TSHR and NM_080426.2 for GNAS). The A of the ATG of the initiator methionine codon is denoted as nucleotide +1.
Data analysis
Dose–response stimulation and competitive binding data were analyzed using GraphPad Prism v5.00 software. For competitive binding studies, a one-site binding model was assumed. Dose–response stimulation analyses were performed by a repeated measured ANOVA followed by the Bonferroni test. Differences in maximum 125I bTSH binding capacity and EC50 were compared using the Mann–Whitney test.
Results
Genetic studies
The propositus was heterozygous for TSHR c.100G > A (Fig. 1, subject II-3), a mutation located in exon 1 that causes a change from glutamic acid (GAG) to lysine (AAG) at residue 34, p.E34K. No mutations were found in the PAX8 gene. The mother and one daughter of the propositus were also heterozygous for p.E34K (Fig. 1, subjects I-2 and III-1).
The propositus's wife and the two daughters were heterozygous for GNAS c.750_751insA, henceforth referred to as GNASMut (Fig. 1 subjects II-4, III-1, and III-2), a mutational insertion located on exon 10 of GNAS that causes a change from asparagine (AAC) to lysine (AAA) at residue 250, p.N250K, and a frameshift in the reading frame with a stop codon, TGA, which is 35 amino acids downstream. The parents and the brother of the propositus's wife were wild type (Fig. 1 subjects I-3, I-4, and II-5).
Dose–response studies
CRE-luciferase activity was dependent on the type of pSVL-TSHR vector transfected: pSVL-TSHR-WT (250 ng/well) >pSVL-TSHR-WT (125 ng/well) plus pSVL-TSHR-E34K (125 ng/well) > pSVL-TSHR-E34K (250 ng/well) (Fig. 2). No differences in basal CRE-luciferase activity were observed (without rhTSH stimulation). EC50 was significantly lower for the wild-type TSH receptor (4.07 ± 0.72 U/L) than for the E34K TSH receptor mutant (13.40 ± 3.98 U/L).
A significantly higher CRE-luciferase response was obtained in cells transfected with a combination of pSVL-TSHR-WT (125 ng/well) and pSVK3-GNAS vectors than in cells transfected with pSVL-TSHR-WT and the pSVK3-GNAS-Mut (125 ng/well), at all rhTSH doses and also basally (without rhTSH stimulation; (Fig. 3). However, the CRE-luciferase response in those cells transfected with the pSVL-TSHR-WT (125 ng/well) combined with pSVK3-GNAS-WT (125 ng/well) was not significantly different to that observed with pSVL-TSHR-WT (125 ng/well) combined with pSVK3-GNAS-WT (62.5 ng/well) and pSVK3-GNAS-Mut (62.5 ng/well), the combination resembling the mutation status of the wife and one daughter of the propositus (Fig. 3).
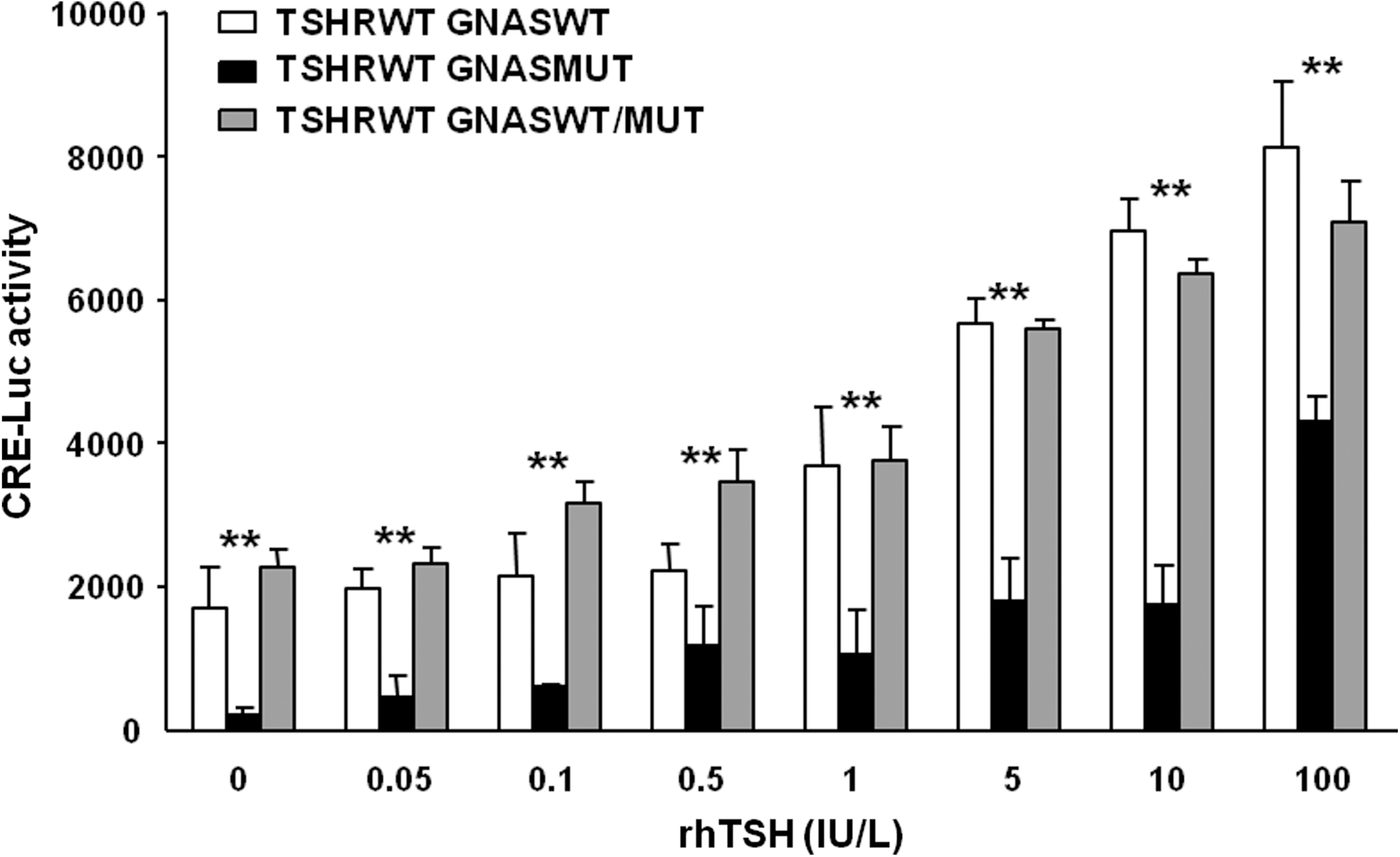
Dose–response curves for CRE-luc activity induced by different doses of rhTSH in COS-7 cells transiently expressing TSHR wild-type and GNAS wild-type and/or the GNASMut mutant. Data are plotted as average ± SE, for two experiments; each point was run in triplicate. Data were analyzed using GraphPad Prism software.
CRE-luciferase response after transfection with a combination of vectors that simulated the genotype of subject III-1, heterozygous for TSHR p.E34K, and GNASMut was significantly lower than that in the wild type (Fig. 4). Cells transfected with pSVL-TSHR-WT (125 ng/well) together with pSVK3-GNAS-WT (125 ng/well) showed a higher cAMP response than those transfected with the combination of pSVL-TSHR-WT (62.5 ng/well), pSVL-TSHR-E34K (62.5 ng/well), pSVK3-GNAS-WT (62.5 ng/well), and pSVK3-GNAS-Mut (62.5 ng/well), at rhTSH concentrations of 5, 10, and 100 U/L (Fig. 4). However, no differences in CRE-luciferase response were observed between cells transfected with pSVL-TSHR-WT (62.5 ng/well) plus pSVL-TSHR-E34K (62.5 ng/well) and pSVK3-GNAS-WT (125 ng/well), regarding the same vector combination plus pSVK3-GNAS-Mut (62.5 ng/well) (Fig. 4).
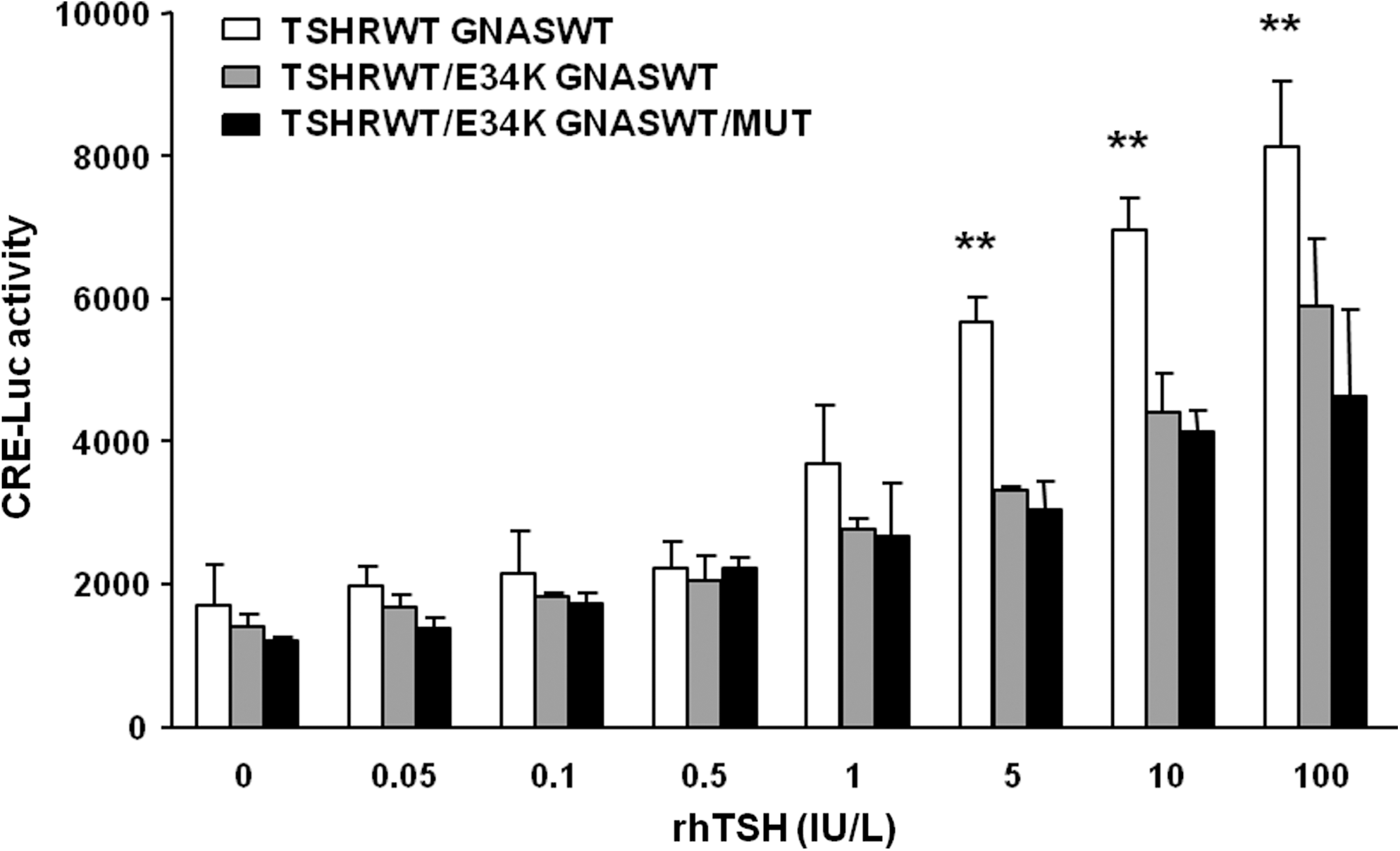
Dose–response curves for CRE-luc activity induced by different doses of rhTSH in COS-7 cells transiently expressing TSHR wild-type and GNAS wild-type versus TSHR wild-type and the p.E34K mutant with GNAS wild-type or GNAS wild-type plus the GNASMut. Data are plotted as average ± SE, for two experiments; each point was run in triplicate. Data were analyzed using GraphPad Prism software.
Binding studies
The TSHR E34K mutant showed 60% of the maximum 125I bTSH binding capacity of the wild-type TSH receptor, and the IC50 was also lower for the E34K (15.03 ± 4.08 IU/L) than the wild-type receptor (1.53 ± 0.38 IU/L) (Fig. 5).
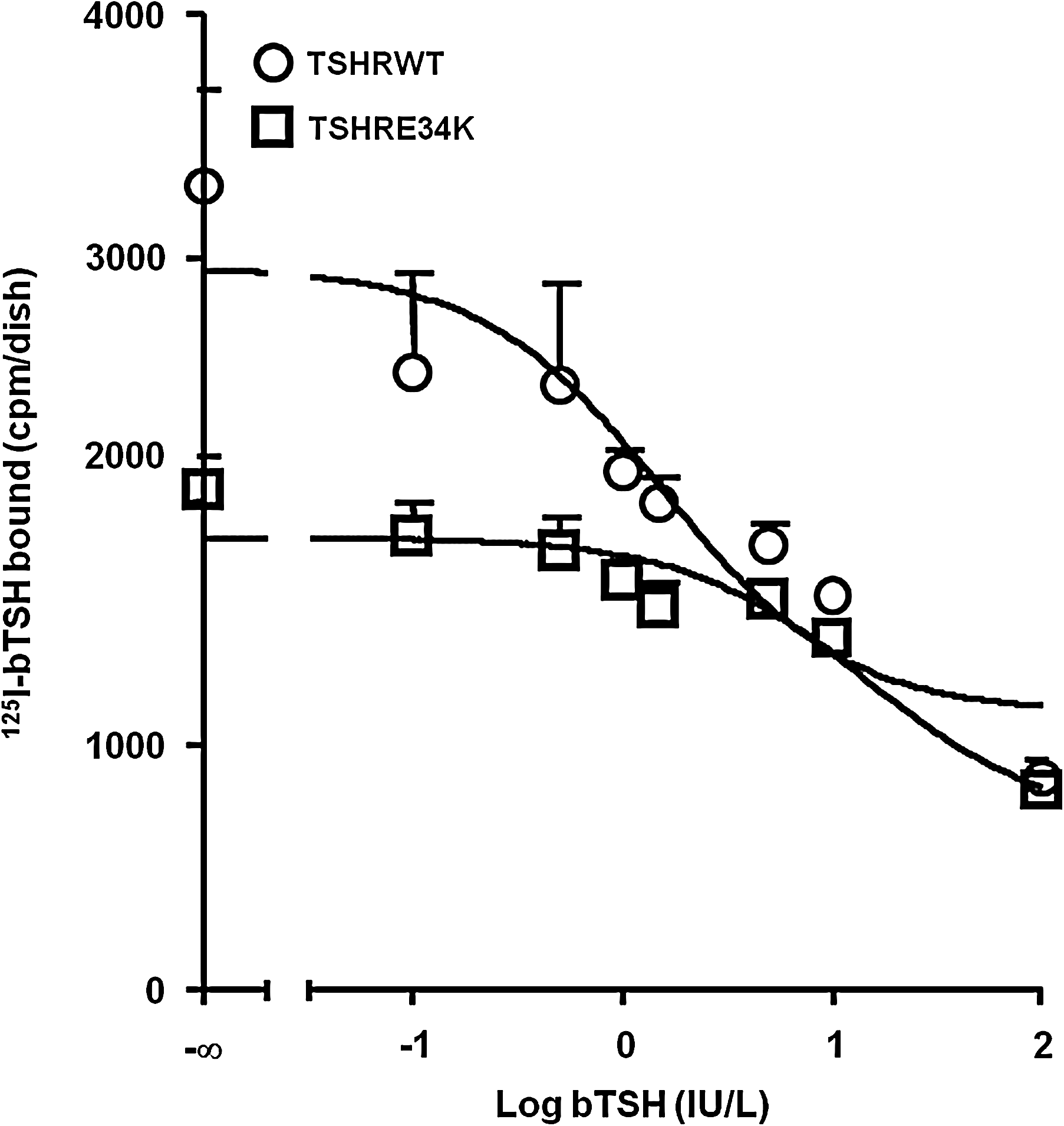
Competitive binding assay showing binding properties of the wild-type TSH receptor and p.E34K mutant, expressed in COS-7 cells and using a 125I-bTSH tracer. The data are from a single representative experiment, with all points run in duplicates, plotted as average ± SE. Results were analyzed using the GraphPad Prism v5.00 software and a one-site binding model. bTSH, bovine TSH.
Discussion
We have reported a family with several members showing TSH resistance caused by combinations of TSHR and GNAS LOF mutations. The propositus was heterozygous for the TSHR mutation p.E34K, located at the extreme N-terminus of the receptor ectodomain, in a cysteine-rich box that contributes to the tertiary structure of the ectodomain (12). p.E34K has been previously reported in heterozygosity in an Italian patient with elevated TSH, normal-low FT4, and marginally elevated FT3, whose father, also heterozygous for p.E34K, was euthyroid (13,14). Functional studies on that patient showed that p.E34K expression at the cell surface was nearly 20% lower, and maximum 125I-bTSH binding 50% lower, than for the wild-type receptor (15); at bTSH doses of 10 and 100 U/L, p.E34K showed a 30% lower cAMP production than the wild-type receptor (14). In our case, the TSHR p.E34K mutant had a 40% lower maximum 125I-bTSH binding, higher IC50, and 60% lower CRE-luciferase activity than the wild-type receptor at high levels of rhTSH stimulation. Taken together, these results suggest that THSR p.E34K has a lower cell-surface expression and lower affinity for TSH than the wild-type TSH receptor.
In the present study, a CRE-luciferase reporter plasmid was used to evaluate the activity of wild-type and mutant TSHR and Gs proteins. The CRE-luciferase reporter is a fast, sensitive, easy, and relatively cheap method to examine the pharmacology of G-protein-coupled glycoprotein (16 –18). Although this method is mainly used to estimate the activity of Gsα-AC-cAMP cascade, it lacks the specificity of direct measurement of cAMP because CREB activation is also induced by increases in intracellular Ca+2 (17). It is likely that the higher doses of rhTSH used in our in vitro experiments stimulated the PLC-IP3-DAG pathway resulting in an increase in intracellular Ca+2 levels and subsequent increase in CRE-luciferase activity. In theory this effect should not represent a major problem in experiments transfecting wild-type and/or LOF mutant TSHR plasmids since an LOF mutation located at the TSH binding site of TSHR will affect both AC/cAMP and PLC pathways. However, it could be a potential problem in the experiments cotransfecting TSHR plasmids with LOF mutant GNASS plasmids, since the exposure to a high dose of rhTSH will increase CRE-luciferase activity through PLC pathways. Under these circumstances, a direct measurement of intracellular cAMP would provide more accurate information than CRE-luciferase activity regarding the effect on cell signaling in GNASS LOF mutants.
CRE-luciferase activity in cells transfected with a combination of both TSHR wild-type and p.E34K mutant vectors was 25% lower than activity observed with the TSHR wild-type vector alone. These results may explain the presence of TSH resistance observed in subjects heterozygous for p.E34K (13), but such a mild decrease in CRE-luciferase activity does not seem to explain the severity of hypothyroidism seen in our propositus, nor the absence of TSH resistance, both in the heterozygous progenitors of our patient and in the progenitors of the previously reported Italian patient (14). Indeed, the mother of our propositus had a history of hyperthyrodism caused by Graves' disease. The possibility that our propositus might have had an additional unknown mutation responsible for hypothyroidism cannot be ruled out. Congenital hypothyrodism can be associated with mutations in PAX8, NK2 homeobox 1 (NKX2.1 or TTF-1), and dual-oxidase 2 (DUOX2) genes (19,20). No mutations in the coding sequence of PAX8 were found in our propositus by direct sequencing. NKX2.1 mutations cause choreoathetosis, neonatal respiratory distress, and CH, and the majority of patients with NKX2.1 present neurological signs that were not present in our propositus (21). Monoallelic DUOX2 mutations can cause mild permanent CH (22,23) with a partial iodide organification defect, and the presence of a DUOX2/DUOXA2 defect cannot be excluded in our patient.
The propositus's children showed physical features suggesting Albright's hereditary osteodystrophy that led us to search for mutations in the GNAS locus. The children's mother was heterozygous for GNASMut, a de novo mutation not present in her own parents; she presented mild features of Albright's hereditary osteodystrophy without PTH and TSH resistance, and her menstrual periods were normal. These findings indicate that in contrast to the children, their mother had a Gsα haploinsufficiency but without thyroid gland imprinting (9).
GNASMut is a severe mutation that is predicted to produce truncated proteins; accordingly, our functional studies showed that cells transfected with a vector-bearing GNASMut showed almost no basal CRE-luciferase activity (i.e., without rhTSH stimulation); at high rhTSH doses (100 U/L) there was a small but significant increase in CRE-luciferase activity that probably resulted from the activation of COS-7 cells endogenous wild-type Gsα and/or of the PLC pathway. However, after cotransfection with both wild-type and mutant GNAS vectors, no differences in CRE-luciferase activity were observed (compared with the wild-type vector alone). These results indicate that when biallelically expressed the GNASS mutant did not interfere with the wild-type GNASS, and that one normal allele is sufficient to maintain an adequate cellular cAMP production. These results also agree with the clinical observation that the propositus's wife was euthyroid but their children showed TSH resistance due to preferential expression of the mutated maternal allele in their thyroid glands.
Our functional studies showed that CRE-luciferase activity in cells transfected with TSHR wild-type plus GNASS wild-type vectors was similar to that observed using the combination of TSHR wild-type plus GNASS wild-type and mutant vectors, but higher than for the combination of TSHR wild-type plus TSHR E34K and GNASS wild-type plus mutant vectors. However, although the child compound heterozygous for TSHR p.E34K and GNAS-Mut had higher TSH levels in neonatal screening than her sister, who was wild-type for TSHR and heterozygous for GNAS-Mut, both children had normal FT4 and FT3 thyroid hormone levels and currently show no clinical signs of hypothyrodism, due to their treatment with thyroid hormone replacement from an early age. Altogether, these observations suggest that, under nonimprinting conditions, the combination of TSHR wild-type plus TSHR E34K and GNASS wild-type plus mutant vectors, as well as the combination of TSHR wild-type plus TSHR E34K and GNASS mutant vectors, is equally effective in causing TSH resistance, pointing then to TSHR E34K as the main cause of this problem; however, the observation that family members heterozygous for TSHR E34K were normo- or even hyperthyroid suggests that the imprinted maternal GNASMut allele was the main cause of TSH resistance in the children and that the contribution of the TSHR p.E34K mutation was probably not sufficient to decrease production of thyroid hormones by the thyroid gland.
In conclusion, we have described a family with several members showing TSH resistance caused by a combination of a heterozygous TSHR mutation, p.E34K, and a previously unreported heterozygous mutation in the GNAS complex locus, c.750_751insA. Our functional studies have shown that the TSHR pE34K mutant, in both homozygosity and heterozygosity, had a lesser capacity to increase CRE-luciferase activity than did the TSHR wild type. In agreement with its predicted production of transcripts codifying for truncated proteins, the pSVK3-GNAS-Mut was found not to be able to activate cAMP production. Moreover, when biallelically expressed, the pSVK3-GNAS-Mut did not have any significant effect on cAMP production, regardless of whether it was transfected with the TSHR wild-type or TSHR p.34K mutant. The differences in clinical phenotype between family members heterozygous for TSHR pE34K mutations remain unexplained. These results also suggest that TSH resistance in the two children, one of whom was wild-type for TSHR and the other heterozygous for TSHR p.E34K, was probably caused by preferential expression in the thyroid gland of the mutated maternal allele, with less clinical contribution from p.E34K.
Footnotes
Acknowledgments
To the members of the family for their willingness to collaborate in the study. This work was supported by the Fondo de Investigaciones Sanitarias FIS (grant PI030401 to JL-A), Ministerio de Educación (grant SAF2006-02542 to JL-A), and Xunta de Galicia (grant PGIDIT04PXIC20801PN to JL-A, PGIDIT06PXIB 208360PR to JL-A).
Disclosure Statement
The authors declare that no competing financial interests exist.