
Abstract
Background:
We have previously described a p.G533C substitution in the
Methods:
Plasmids expressing RET mutants (p.G533C and p.C634Y) and RET wild type were stable transfected into a rat thyroid cell line (PCCL3). Biological and biochemical effects of RET p.G533C were investigated both in vitro and in vivo. Moreover, we report the first case of pheochromocytoma among the RET p.G533C–carriers in this Brazilian family and explore the RET mutational status in DNA isolated from pheochromocytoma.
Results:
Ectopic expression of RET p.G533C and p.C634Y activates RET/MAPK/ERK pathway at similar levels and significantly increased cell proliferation, compared with RET wild type. We additionally show that p.G533C increased cell viability, anchorage-independent growth, and micronuclei formation while reducing apoptosis, hallmarks of the malignant phenotype. RET p.G533C down-regulates the expression of thyroid specific genes in PCCL3. Moreover, RET p.G533C-expressing cells were able to induce liver metastasis in nude mice. Finally, we described two novel RET variants (G548V and S556T) in the DNA isolated from pheochromocytoma while they were absent in the DNA isolated from blood.
Conclusions:
Our in vitro and in vivo analysis indicates that this mutation confers a malignant phenotype to PCCL3 cells. These findings, in association with the report of first case of pheochromocytoma in the Brazilian kindred, suggest that this noncysteine mutation may be more aggressive than was initially considered.
Introduction
Germ-line mutations in RET gene have been identified as the underlying cause of the multiple endocrine neoplasia type 2 (MEN 2) syndrome, an inherited autosomal dominant disorder with high penetrance. MEN 2 syndrome includes three different clinical variants. MEN 2A is characterized by the presence of medullary thyroid carcinoma (MTC), pheochromocytoma, and hyperparathyroidism. MEN 2B is characterized by early onset of MTC, pheochromocytoma and, more rarely, by ganglioneuromas, thickened cornea nerves, and marfanoid habitus. Familial MTC (FMTC) is characterized by MTC as the only clinical feature, although the accurate distinction between FMTC and MEN 2A is not always clear [reviewed by refs. (2,3)].
The clinical variability observed in MEN 2 syndrome reflects genetic heterogeneity. Most mutations associated with MEN 2A and FMTC were identified in cysteine residues in the extracellular domain (exons 10 and 11). Almost all individuals with MEN 2B have an identical mutation in codon 918 (exon 16) (4), although A883F mutation (exon 16) has been described in a few cases (5).
Recently, the American Thyroid Association created specific MTC clinical guidelines that combined the MTC literature with evidence-based medicine and the knowledge of a panel of expert clinicians. The guideline recommendation on the timing of prophylactic thyroidectomy and the extent of surgery is based in this model that utilizes genotype-phenotype correlations to stratify mutations into four (A–D) risk levels (6).
Although it is established that RET mutation in MEN 2 causes RET constitutive activation and that there is a clear genotype-phenotype correlation, in vitro studies have demonstrated differences in transforming activity of RET receptor carrying different mutations. Additionally, for less common mutations, controversy still exists regarding the timing and extent of surgical intervention. Hence, functional analysis could provide further evidence for the transforming potential of these mutations.
We have previously described a mutation in exon 8 of the RET gene, which leads to a substitution of p.G533C in a family with MTC as the only clinical feature (7). This mutation was later identified in two Greek families with FMTC (8). Additionally, the p.G533C mutation was described in unrelated Greek families with pheochromocytoma as the first clinical manifestation (9,10). Follow-up of these patients has shown high levels of basal and stimulated serum calcitonin levels. Total thyroidectomy was performed, and histological examination confirmed the presence of MTC. The occurrence of MTC and pheochromocytoma in these Greek kindred provide the first evidence that p.G533C mutation leads to the MEN 2A phenotype (9,10).
We here functionally explored the transforming potential of RET p.G533C mutation. We also report one patient with pheochromocytoma in this large Brazilian kindred and assessed the mutational status of RET in pheochromocytoma tissue.
Materials and Methods
Pathological data
We previously described a six-generation family with RET p.G533C mutation and MTC as the only clinical manifestation. Out of 76 p.G533C-carriers, 29 had MTC, and 6 CCH (7). Further, we reported new cases harboring RET p.G533C mutation who had MTC before referral to our institution for genetic screening (11). Herein, pathological TNM (pTNM) classification at the time of surgery and the presence of local and distant metastases during follow-up is described in those for whom follow-up data were available (n = 34).
Cell culture
A differentiated thyroid follicular cell derived from 18-month-old Fischer rat (PCCL3) was maintained in medium consisting of Ham F-12 supplemented with 5% FCS (Invitrogen Corp., Carlsbad, CA) and a four hormone mixture (4H) including thyrotrophin (1 mU/mL), hydrocortisone (10 nM), insulin (10 μg/mL), and apo-transferrin (5 μg/mL). A human MTC cell line (TT) was maintained in Ham F-12 supplemented with 10% FBS.
Plasmid constructs and transfection assays
The expressing vector pcDNA3.1 encoding the short RET wild type (Wt) and RET p.C634Y isoforms were kindly donated by Dr Massimo Santoro. The RET p.G533C (mutant) was generated from the RET Wt by site-directed mutagenesis using the QuickChange XL mutagenesis kit according to the manufacturer's recommendations (Stratagene, La Jolla, CA). The primers were 5′ AGGAGTGTGGCTGCCTGGGCTCCC 3′ (sense) and 5′ GGGAGCCCAGGCAGCCACACTCCT 3′ (antisense). The mutations were confirmed by direct sequencing using the Big DyeTerminator Cycle Sequencing Ready Reaction Kit and analyzed in the ABI PRISM 3100 genetic analyzer (Applied Biosystems, Foster City, CA) as described (12).
About 10 μg of each construct were stably transfected into PCCL3 cells by electroporation (Bio-Rad Laboratories, Inc., Hercules, CA). For each transfection experiment, gentamicin-resistant colonies (300 μg/mL) were pooled and checked for ectopic expression of RET by quantitative polymerase chain reaction (qPCR). To this end, total RNA was isolated and reverse transcribed to cDNA. An aliquot of cDNA was used in 20 μL PCR reaction containing SYBR Green PCR Master Mix (Applied Biosystems) and 200 nM of each primer for target (RET) or reference gene (Actb) as described (13). qPCR reactions were performed in triplicate, the threshold cycle (Ct) was obtained using Applied Biosystem software and was averaged (SD ≤ 1). Gene expression was calculated as described (13,14). Primer sequences are described in Supplementary Table S1 (Supplementary Data are available online at
Protein analysis
Cells expressing RET mutants and Wt were serum- and H4-starved overnight. Total cell lysates were prepared as described (15). For immunoprecipitation experiments, 2 mg of protein was incubated with anti-RET (Cell Signaling, Beverly, MA) at 4°C, and immunocomplexes were recovered by adsorption to protein A/G plus-agarose beads (sc-2003, Santa Cruz Biotechnology) at 4°C overnight. Total protein (50 μg) or imunocomplexes were electrophoresed on 4%–12% SDS-polyacrylamide gel (Invitrogen Corp.), followed by Western blot analysis as described (15). The membrane was incubated overnight at 4° C with antiphospho-ERK (pERK) (1:1000), anti-ERK (1:500), anti-RET (1:500) (Cell Signaling), or antiphospho-RET Y1062 (1 μg/mL; R&D Systems, Inc., Minneapolis, MN). Goat anti-rabbit secondary antibody (HRP conjugated) was used at 1:5000 dilution (DAKO, Glostrup, Denmark). The immune complexes were detected using using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL). Band intensities were quantified using Molecular Dynamics Image Quant System (GE Healthcare, Buckinghamshire, United Kingdom).
Cell proliferation assay
Cell proliferation rate was measured with MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay as described (16). In brief, PCCL3 cells (104) expressing RET mutants or Wt were seeded in quintuplicate. At each time point, cells were incubated with MTT reagent for 3 hours. Absorbance was measured at a wavelength of 560 nm.
Cell viability and apoptotic assays
For cell viability, PCCL3 cells (104) were seeded in quintuplicate. At the indicated time, cells were stained with Guava ViaCount reagent (Guava Technologies, Hayward, CA). Viable and nonviable cells were differentially stained with Guava ViaCount reagent and counted by a Guava Mini flow cytometer according to the manufacturer's specifications (Guava Technologies).
To detect apoptosis, the cells were double stained with annexin-V and 7-amino-actinomycin D (7-AAD), according to the manufacturer's recommendations (Guava Nexin method, Guava Technologies). Both early apoptotic (annexin-V–positive) and late apoptotic (annexin-V– and 7-AAD–positive) cells were included in the analysis. Results were expressed as the percentage of apoptotic positive cells.
Assay for micronuclei
Cells (7 × 103) were seeded in slide desk (Nunc, Roskilde, Denmark). After 4 days, cells were fixed and stained using rapid panoptic LB (Laborclin, São Paulo, Brazil). Micronuclei were counted under a light microscope at a total magnification of ×400 according to standard procedures (17).
Anchorage-independent growth
Anchorage-independent growth was assessed by a double-layer soft agar assay as described (15). After 4 weeks, cells were stained with MTT (1 mg/mL) and photographed. Colonies formed in soft agar (≥64 cells) were counted from at least four different areas. Two independent experiments were performed in triplicate. The TT cell line that harbors the RET p.C634W substitution was used as positive control.
Expression of genes potentially modulated by RET p.G533C
To investigate whether the RET p.G533C alters the expression of thyroid specific genes in PCCL3 cells, as a consequence of its transforming potential, we next investigated the expression of genes associated with differentiated thyroid function by qPCR: sodium iodide symporter (Slc5a5), thyroid peroxidase (Tpo), thyroglobulin (Tg), and thyroid stimulating hormone receptor (Tshr). We additionally investigated the expression of genes previously described as induced (Cathepsin B (Ctsb) and Decorin (Dcn)) or repressed (Chemokine (C-X-C motif ) ligand 12 (CxcL12) and TIMP metallopeptidase inhibitor 3 (Timp3)) by MEN 2A (p.C634R) and/or MEN 2B (p.M918T) mutations (18).
Total RNA isolation, cDNA synthesis, and PCR reaction were performed as previously mentioned. Gene expression was calculated, as described (19). Primers are described in Supplementary Table S1.
Tumors xenografts in nude mouse
Seven- to 8-week-old female athymic nude (nu/nu) mice were maintained according to the guidelines of the Division of Animal Resources at the Federal University of São Paulo. To evaluate the tumorigenic potential of RET mutant, cells (6 × 106) expressing RET p.G533C and parental cells were injected subcutaneously sc into the flank of each mouse (n = 7) for each experimental group. Mice were then monitored over a 7
Identification of PCCL3 cells expressing RET p.G533C in liver metastasis
To provide evidence that cells expressing p.G533C were able to survive in the circulation, reach the liver, and proliferate into tumor colonies, RET expression was investigated in mouse liver by reverse transcription-polymerase chain reaction (RT-PCR). To this end, total RNA was isolated from liver tissues, reverse transcribed into cDNA, and amplified by PCR as described (13). The identity of the PCR product was confirmed by sequence analysis (11,20). Additionally, the expression of thyroid-specific gene (Tg) was evaluated in liver specimens from control group (n = 2) or nude mice injected cells expressing RET p.G533C (n = 2) by qPCR as previously mentioned. The primers for target gene (Tg) and internal control (Actb) are summarized in Supplementary Table S1.
DNA isolation and RET mutational screening
To investigate whether an additional mutation within RET oncogene could be implicated in the tumorigenesis of pheochromocytoma, genomic DNA was isolated from adrenal tumor using standard methods. Moreover, two MTCs samples obtained from RET p.G533C–carriers were investigated for the presence of RET mutations. PCR products of exons 8–16 of RET gene (GenBank #AJ243297) were submitted to direct sequencing as described (11). Each sample was sequenced at least twice and in both directions.
Statistical analysis
In vitro results and qPCR values were log transformed and submitted to ANOVA test with Student-Newman-Keuls adjustment. Differences yielding a p < 0.05 were considered significant.
Results
Pathological features
Herein we reassess pathological features of 34 RET p.G533C–carriers with MTC whom follow-up data were available. Patients were classified according to pTNM system. Eighteen patients were classified into stage 1, 4 into stage II, and 12 into stage III (Table 1). Although longer follow-up is needed, these results pointed out that the phenotype of MTC in patients with this noncysteine mutation may be more aggressive than originally thought.
Numbers are according to the pedigree of the six-generation family previously published (7).
All patients underwent surgery between 2001 and 2004, and the most recent follow-up was performed between 2009 and 2010.
NA, not available; ND, not detailed; ML, number of metastatic lymph nodes; LD, number of lymph nodes dissected.
Moreover, in our previous study, we reported that two family members died from advanced MTC with metastasis to lung and liver at the ages of 53 and 60, and MTC or CCH was diagnosed in four patients before the age of 18 (7,11).
Forced expression of RET p.G533C in PCCL3 cells
To investigate the transforming potential of RET p.G533C mutation in a thyroid follicular cell, constructs encoding RET mutants (p.G533C or p.C634Y) or RET Wt were permanently transfected into PCCL3 cells. To avoid the interference of clonal selection, clones from each transfectant were pooled together. Expression of the exogenous RET mRNA was confirmed by qPCR. Our results demonstrated that the expression of RET was similar in all transfectants. Moreover, sequencing analysis of PCR products confirmed the presence of RET mutants or RET Wt (data not shown). Pooled cells were used in all in vitro and in vivo assays.
RET p.G533C increases RET and ERK phosphorylation and cell proliferation
We next investigated the ability of p.G533C mutation to convert RET into an oncogene and to stimulate the RET/MEK/ERK cascade. Seeing that several groups have demonstrated that the tyrosine residue 1062 (Y1062) is essential for the transforming activity of RET mutants, we next investigated the phosphorylation level of Y1062 and p42/44 extracellular signal-regulated kinase (pERK 1/2). On starvation, both RET and ERK phosphorylation were higher in mutants (i.e., p.G533C and p.C634Y) than observed in RET Wt. The results are graphically represented (Fig. 1A).
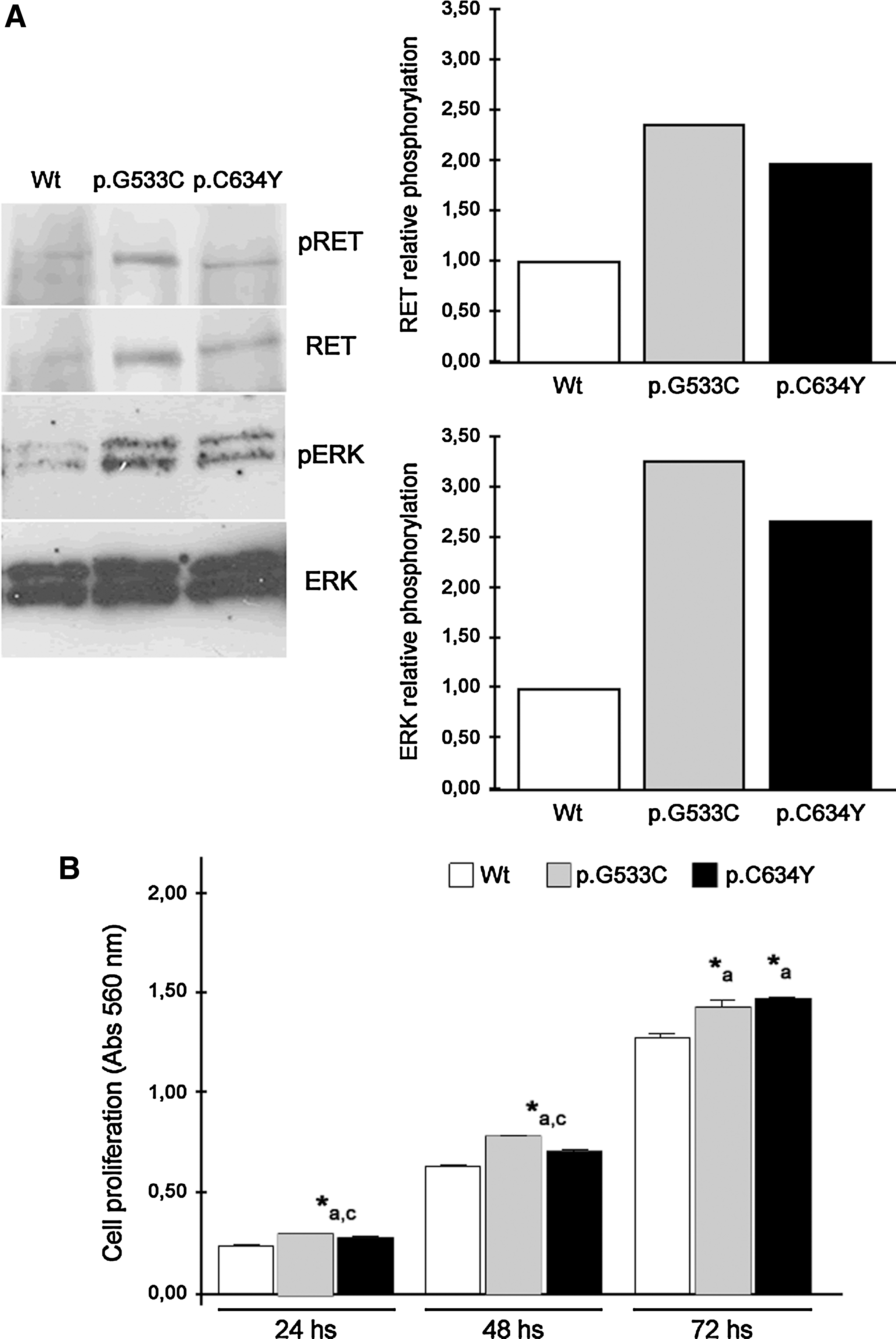
Levels of
Forced expression of RET p.G533C resulted in a significant increase in cell proliferation, when compared with RET Wt. Although at lower levels, ectopic expression of RET p.C634Y increased cell proliferation in comparison with RET Wt (p < 0.05; Fig. 1B). These findings are in agreement with those obtained for pRET and pERK.
RET p.G533C increases cell viability while reduces apoptosis rate of PCCL3 cells
After 24 hours, the expression of RET p.G533C and Wt resulted in a significant increase in cell viability and in a decrease in the apoptosis rate, when compared with parental cells. After 48 hours, the expression of RET p.G533C increased cell viability and reduced the rate of apoptosis when compared with RET Wt and parental cells (p < 0.05; Fig. 2A, B).

Effects of RET p.G533C expression in cell viability, apoptosis, and micronuclei formation.
RET p.G533C induces micronuclei formation in PCCL3 cells
We observed that RET p.G533C stable expression significantly increases the number of cells with micronuclei when compared with parental cells (2.4-fold) or RET Wt (2-fold), suggesting that the increased genomic instability observed in cells expressing p.G533C may be associated with tumor progression. The results are graphically represented (p < 0.01) (Fig. 2C).
RET p.G533C effects on anchorage-independent growth
We noted that RET p.G533C significantly increased the ability of PCCL3 cells to form colonies in semisolid medium, when compared with PCCL3 cells expressing RET Wt (p < 0.05; Fig. 3). The human MTC cell line (TT), which not only displays high clonogenicity ability but also forms large colonies in soft agar, was used as a positive control.
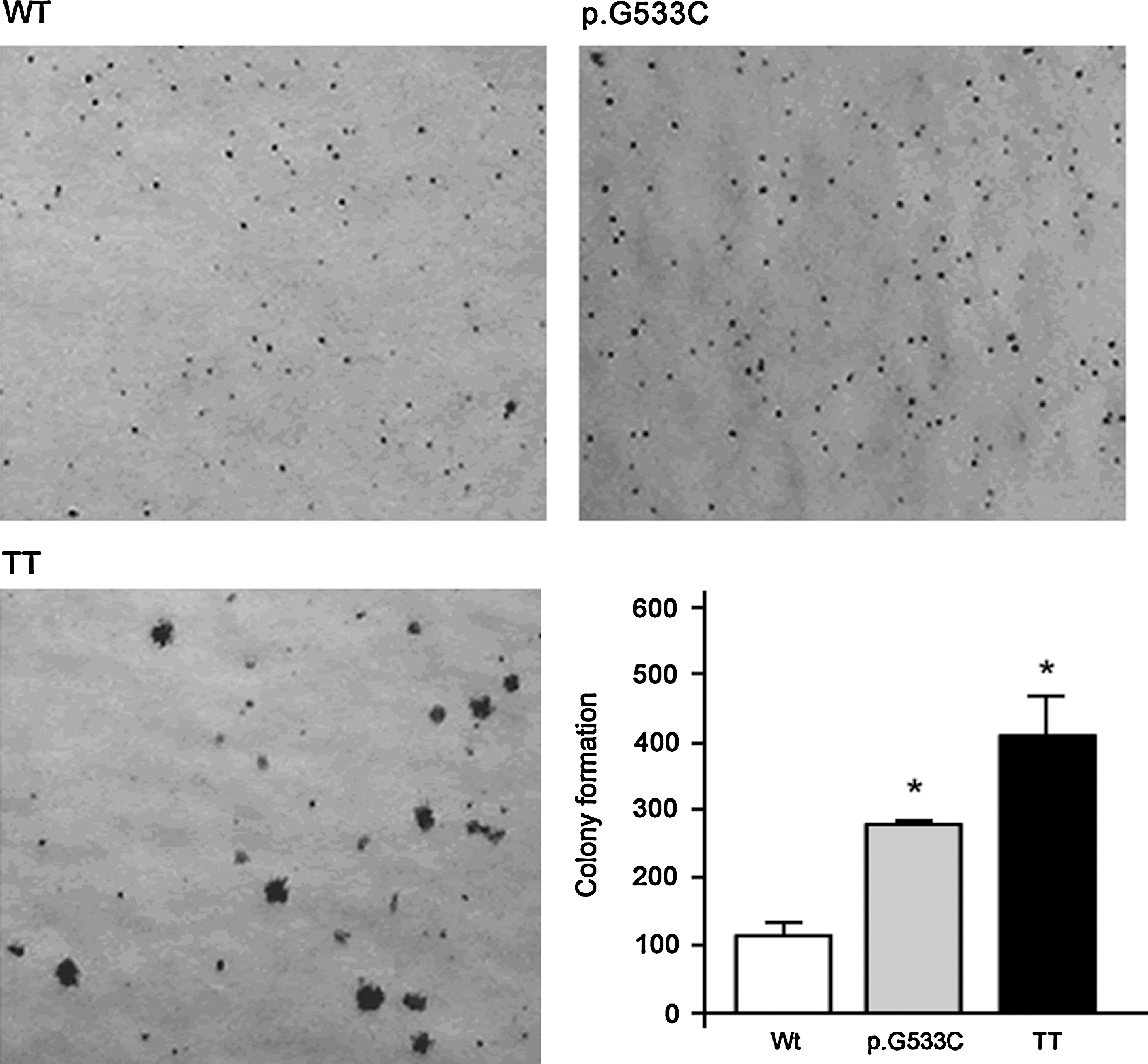
Effect of RET p.G533C expression in anchorage-independent growth. Colonies larger than 64 cells were counted after 4 weeks. A representative picture of RET-expressing cells and positive control (TT cells) is shown. Bar graph shows the mean ± SD of two independent experiments performed in triplicate (*p < 0.05).
RET p.G533C reduces expression of thyroid specific markers in PCCL3 cells and modulates expression of Dcn, Cxcl12, and Timp3
It has been reported that alterations along the RET/RAS/RAF/MAPK pathway promote loss of the differentiated phenotype, measured by the expression of thyroid-specific genes (21). We next investigated whether RET p.G533C affect the expression of thyroid specific genes. The expression of Slc5a5 (11.3-fold), Tpo (10.9-fold), and Tg (3.1-fold) was lower in RET p.G533C expressing cells (p < 0.05), compared with parental cells. Although not significant, we also noted that the expression of Tshr in RET p.G533C expressing cells was lower than the expression observed in parental cells or cells expressing RET Wt (Fig. 4A).
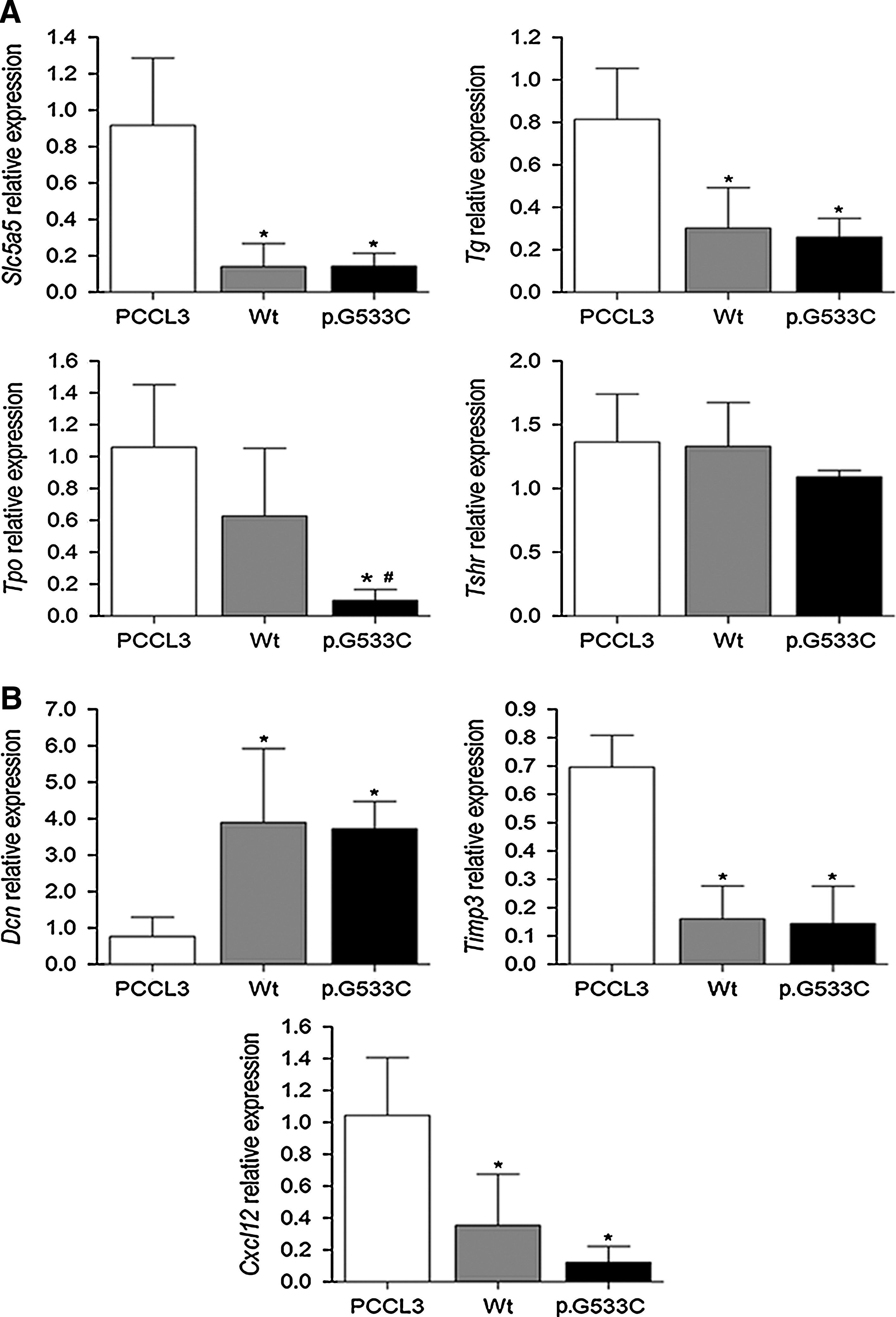
RET modulates the expression of
Identification of genes modulated by RET p.G533C mutation may help better understand the underlying mechanism by which p.G533C induces the transformed phenotype. Similar to those previously reported for RET p.C634R and p.M918T mutations, p.G533C expressing cells induced the expression of Dcn (4.9-fold) while reduced the expression of Timp3 (4.9-fold) and Cxcl12 (5-fold), when compared with parental cells (p < 0.05) (Fig. 4B). Ctsb was expressed at higher levels in p.G533C expressing cells; however, the difference was not statistically significant (data not shown). Although the differences in signal transduction activated by different RET mutations still remains unknown, we here described genes that are modulated by p.G533C in vitro.
Evidence that RET p.G533C–expressing cells were able to induce metastatic dissemination in nude mice
Since anchorage-independent growth is a strong indicator of the transformed phenotype, we next studied the tumorigenic potential of PCCL3 cells expressing RET p.G533C in vivo, and compared them with PCCL3 parental cells. Although nude mice injected with PCCL3 expressing RET p.G533C (n = 7) and parental cells (n = 6) did not form any visible tumors for up to 7 weeks, histological analysis showed the presence of large lesions inside liver of animals injected with RET p.G533C, whereas no lesion was observed in control group. Intriguingly, in all sections examined, not only the liver architecture was disturbed but also several foci of arrested cells could be seen close to the portal vessels (Fig. 5A–C). These findings suggest that both cell extravasation and cell infiltration into the liver parenchyma occurred.
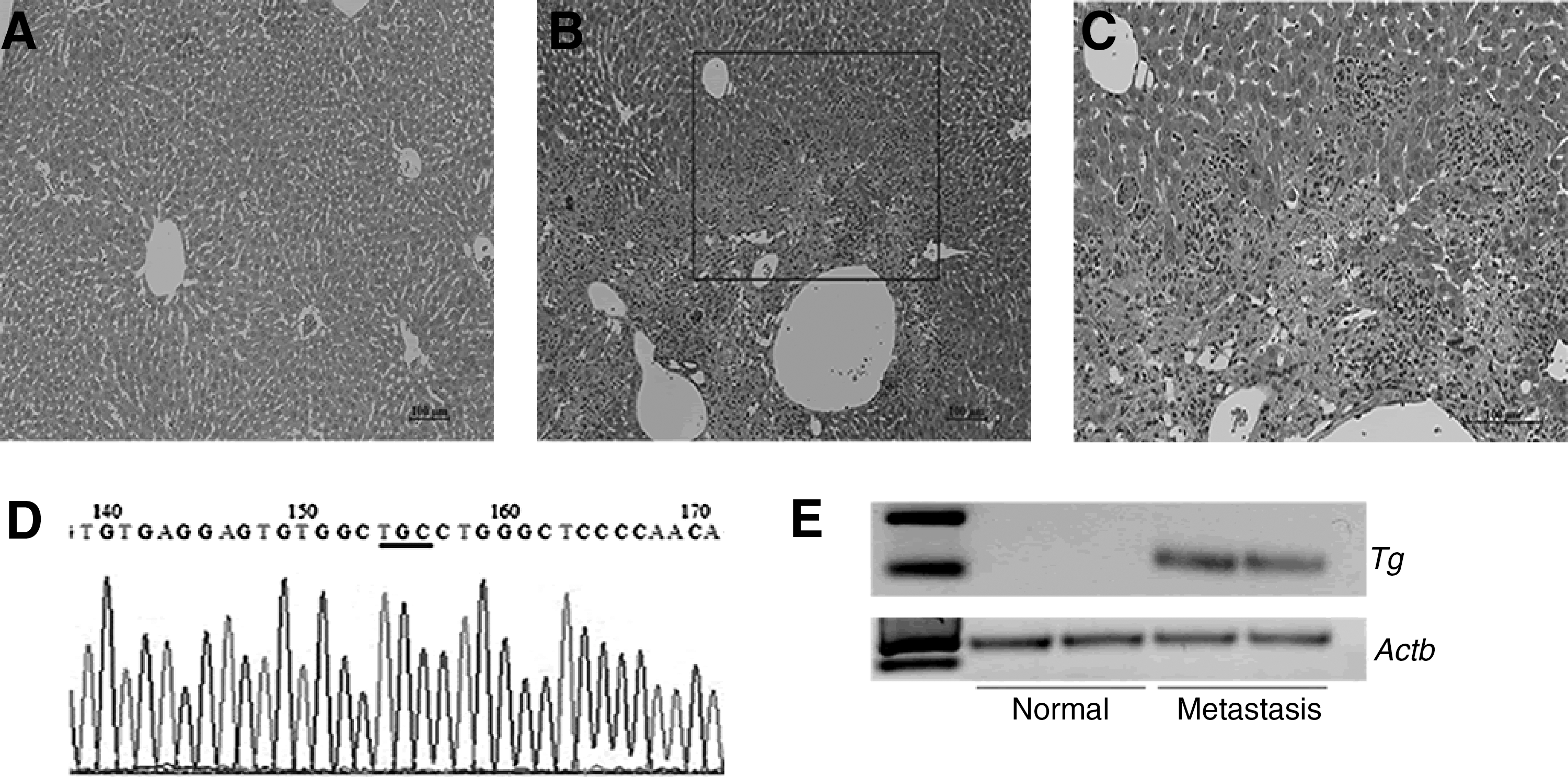
Representative image of
To test whether the PCCL3 cells expressing p.G533C were able to reach the liver and form metastasis, we investigated the expression of the human RET in liver of both experimental and control groups. RT
We next confirmed there was expression of Tg in the liver of animals injected with PCCL3 cells expressing p.G533C, whereas Tg was not expressed in liver of control group (Fig. 5E).
Diagnosis of pheochromocytoma in a RET p.G533C–carrier
A 58 year-old woman was referred to the Endocrinology Service of Hospital das Clínicas, Federal University of Minas Gerais, for evaluation of a thyroid nodule (0.8 cm). Serum calcitonin level was 188 ng/mL (normal values <10 ng/mL). Laboratory tests excluded hyperparathyroidism and pheochromocytoma, as calcium concentrations and urinary metanephrines levels were normal. Total thyroidectomy with central lymph node dissection was performed. Pathological findings showed MTC with lymph node metastasis. At the age of 63, the patient had a hypertensive crisis. Magnetic resonance imaging (MRI) showed a large mass in the right adrenal gland measuring 4.3 × 1.7 cm. An intermediate signal on T1 and high signal on T2-weighted was identified, suggesting pheochromocytoma. Urinary metanephrines were normal; normetanephrine: 0.40 (reference level <0.41 mg/24 hours) and metanephrine: 0.11 (reference level <0.31 mg/24 hours). The patient was referred for laparoscopic right adrenalectomy, and histopathology examination confirmed the diagnosis of pheochromocytoma (case 7, Table 1).
Identification of novel RET variants in pheochromocytoma
In addition to the RET p.G533C mutation, two novel variants within RET oncogene were found in DNA isolated from pheochromocytoma: a G
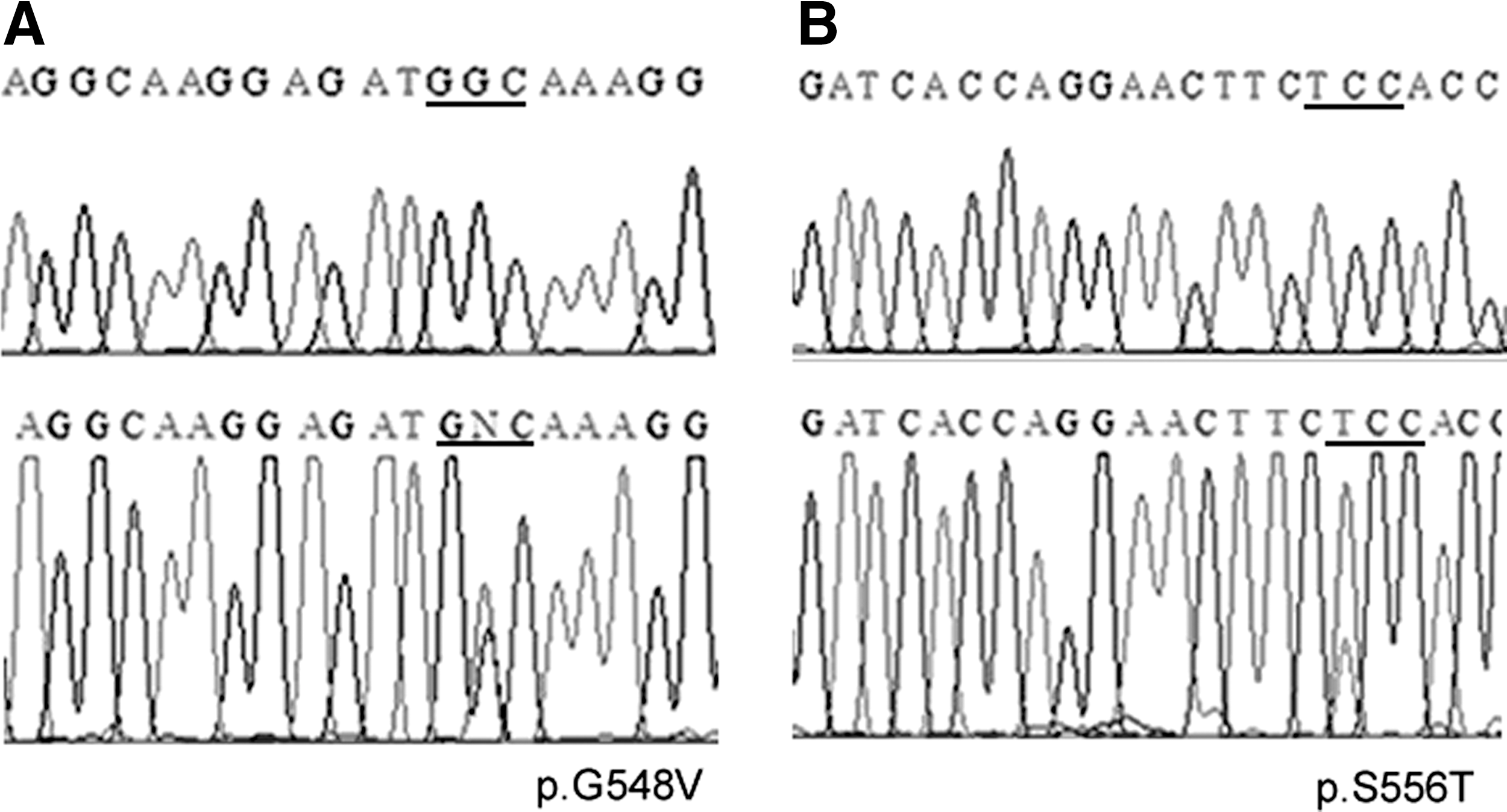
Two novel variants
Discussion
In 2003, our group described a missense mutation in exon 8 of RET gene in a six-generation family with MTC as the only clinical feature (7). This mutation, which leads to a p.G533C substitution in the cysteine-rich domain of RET, was later reported in two Greek families with FMTC (8).
Subsequently, it was reported that two unrelated Greek families harboring RET p.G533C mutation had pheochromocytoma as the first clinical manifestation (9,10). Additionally, the index cases in the reported Greek families had a mild, late form of MTC. These findings, associated with the fact that none of family members died from MTC-related causes, led the authors to suggest that RET p.G533C mutation is associated with a milder phenotype of the MEN 2A (9,10). Further, all mutations identified within exon 8 of RET oncogene were classified by the American Thyroid Association management guidelines for patients with MTC as risk level A, which is the lowest risk level for aggressive MTC (6).
Recently, a new mutation (p.C515S) within exon 8 of RET oncogene was described (22). Functional analysis showed that this mutation was unable to induce foci formation in NIH-3T3 cells, suggesting a reduced oncogenic potential when compared with MEN 2A mutants. When this manuscript was in the final stage of preparation, Muzza et al., described three new variants within exon 8 of RET oncogene (23). The identified variants (p.A510V and p.E511K and p.C531R) had higher transforming potential than RET Wt, but were significantly lower than RET p.C634R. Importantly, the p.E511K mutation was associated with a slight increase in transforming potential in vitro, but it was associated with distant metastasis and, therefore, with a more aggressive clinical behavior. The p.A510V mutation was associated with an aggressive phenotype with persistent hypercalcitoninemia. Altogether, these findings not only highlight the fact that the prevalence of exon 8 mutations are higher than previously thought, but also that they can be associated with either FMTC or MEN 2A and have different biological behavior.
Here, we explored the biological function of p.G533C mutation in PCCL3 cells. RET p.G533C had similar levels of RET Y1062 and ERK1/2 phosphorylation, when compared with that of RET with codon 634 mutation (p.C634Y). Importantly, RET mutants showed higher levels of RET Y1062 and ERK1/2 phosphorylation, compared with RET Wt. Although further analysis is needed, these findings suggest that p.G533C and p.C634Y may have similar signaling abilities.
Given that RET/MEK/ERK signaling pathway is an important intracellular cascade that leads to cell proliferation and survival, we next investigated whether RET p.G533C alters cell proliferation. Consistent with RET and ERK1/2 phosphorylation results, p.G533C and p.C634Y increased cell proliferation when compared with RET Wt, mainly after 72 hours.
Altogether, these results suggest that the p.G533C mutation has effects similar to that observed for p.C634Y substitution. Investigating the role of other tyrosine residues in the kinase domain will help better understand the biochemical and biological proprieties of RET p.G533C mutant.
We next showed that p.G533C significantly increased cell viability, while reducing apoptosis.
It has been demonstrated that transformation of thyroid PCCL3 cells with genes coding for effectors along the MAPK pathway (i.e., RAS and BRAF) induces genomic instability (24,25). These findings, along with the observation that MEN 2-associated tumors from the same patient can have different genetic alterations, raised the possibility that RET p.G533C could induce micronuclei formation, as a consequence of genomic instability. We demonstrated that RET p.G533C significantly increases the number of cells with micronuclei, suggesting that the expression of RET mutant in PCCL3 cells may promote tumor progression by increasing genetic instability.
Although it formed less and smaller colonies in semisolid medium than the positive control, we noted that p.G533C formed more colonies in soft agar than Wt. These findings suggest that p.G533C triggers a more invasive phenotype than Wt.
In the current study, in vivo characterization showed that RET p.G533C cells were not able to form visible tumors in nude mice. Similar results were also found by Santoro et al. (26). However, we observed a widespread infiltration of PCCL3 cells expressing RET p.G533C within the liver. This result, in association to the colony formation assay, suggests that the cells may be capable of surviving without attaching to a substratum, a prerequisite for metastasis. RT-PCR analysis with subsequent DNA sequencing of the PCR product and expression of thyroid-specific gene in the liver confirmed that the liver-infiltrating cells expressed human RET p.G533C. Of note, in patients with MTC, the liver is a major site of metastatic disease.
Interestingly, metastatic colonies in the liver were mainly located near the main blood vessels of the liver portal vein. Importantly, the more metastatic cells, the greater the likelihood that these cells will be resistant to various therapeutic modalities. Therefore, screening of potential RET inhibitors for RET p.G533C is required.
Although clinical and laboratory follow-up reveal that the several RET p.G533C–carriers had a benign course and became disease-free, 12 of 34 had lymph node metastases or invasion at the time of diagnosis, two family members died from metastatic disease to lung and liver, and MTC or CCH was diagnosed in four patients before age of 18 years.
We also sought to determine whether RET p.G533C could affect the expression of thyroid specific genes in vitro, as a consequence of transformed phenotype. RET p.G533C, similarly to RET Wt, reduces the expression of thyroid specific genes and, therefore, may play a role in reversing the differentiated state of PCCL3 cells. It is worth mentioning that the expressions of Tpo and Tshr were lower in cells expressing p.G533C when compared with those expressing RET Wt.
To further elucidate how MEN 2 mutations promote tumorigenesis, we next investigated the expression of genes previously identified as differentially expressed in NIH3T3 cells expressing MEN 2A and MEN 2B mutant proteins in comparison with parental cells (18). Similar to p.C634R and p.M918T mutants, RET p.G533C induced the expression of Dcn while reduced the expression of Timp3. However, a similar effect was observed in cells expressing RET Wt. These findings suggest that exogenous expression of RET may modulate the expression of a number of genes. Regarding Cxcl12, it has been demonstrated that ectopic expression of RET/PTC1 rearrangement triggers the expression of inflammation-invasion related genes such as CXCL12 (27). Intriguingly, we described a reduced expression of Cxcl12 with expression of the RET mutant. Our findings are comparable with those of Watanabe et al., who verified that RET p.C634R and p.M918T mutants repressed the expression of Cxcl12 (18).
In the current study, we also report a 63-year-old woman in whom pheochromocytoma was diagnosed during follow-up. Although RET p.G533C have been previously associated with MEN 2A phenotype in Greek kindred (9,10), our finding reinforces the need for screening for pheochromocytoma and primary hyperparathyroidism in p.G533C-carriers.
Interestingly, in unrelated Greek families, pheochromocytoma was the presenting feature, whereas in our family it was reported in only 1 out of 76 p.G533C-carriers. These findings suggest that the pheochromocytoma penetrance may differ between Brazilian and Greek families. However, the molecular mechanisms associated with this variability in clinical expression are poorly understood. It has been suggested that a second hit which causes a dominant effect of the mutant RET allele may be a potential mechanism for pheochromocytoma tumorigenesis in patients with MEN 2A (28). We here described two novel variants (p.G548V and p.S556T) within RET oncogene in pheochromocytoma that were absent in the blood cells from the same patient and in MTC from two patients with RET p.G533C mutation. Consistent with previous studies (28), we provide additional evidence that additional genetic events may be associated with tumorigenesis of MEN 2A-associated pheochromocytoma. Although further analysis is needed to demonstrate the transforming potential of these variants, it highlights the need for screening for mutations in non-“hot spot” regions.
In summary, we functionally explore the biological and biochemical effect of the RET p.G533C mutation in PCCL3 cells. Overall, our in vitro and in vivo analysis indicates that this mutation appears to confer more malignant properties than was initially thought. We also describe one patient with pheochromocytoma in a large Brazilian kindred and identified two novel RET variants in DNA isolated from the pheochromocytoma. Longer follow-up of the patients and further studies on signaling pathways activated by RET p.G533C, p.G548V, and p.S556T mutations, in comparison with other noncysteine and cysteine RET mutants, will provide new insights into the mechanism associated with the pathogenesis of p.G533C-MEN 2A associated tumors.
Footnotes
Acknowledgments
This work was supported by The São Paulo State Research Foundation (FAPESP) (grants 05/60330-8, 06/60402-1 and 09/11257-7). J.M.C. and R.M.B.M. are investigators of the Brazilian Research Council (CNPq). M.N.L.O., F.R.M.L., A.U.B. and J.P.H. are scholars from FAPESP. R.T. is scholar from CAPES.
Disclosure Statement
The authors declare that there is no conflict of interest which could be perceived as prejudicing the impartiality of the research reported.