
Abstract
Background:
Graves' ophthalmopathy (GO) and lymphedema share some pathogenetic mechanisms, such as edema, inflammation, and adipogenesis. The aim of this study was to examine similarities and differences between chronic GO and chronic lymphedema.
Methods:
Intraorbital adipose tissue was collected from patients with active (n = 10) or chronic GO (n = 10) and thyroid-healthy controls (n = 10). Arm subcutaneous adipose tissue was obtained from patients with chronic arm lymphedema (n = 10), where the unaffected arm served as a control. Gene expression was studied using microarray and real-time polymerase chain reaction.
Results:
The following genes were significantly upregulated (p < 0.05) in lymphedema but not in GO and have functions in wound healing, fibrosis, fat metabolism, inflammation, differentiation, development, adhesion, and the cytoskeleton: ATP-binding cassette, sub-family G (WHITE), member 1 (ABCG1), actin, alpha 2, smooth muscle, aorta (ACTA2), secreted frizzled-related protein 2 (SFRP2), tenascin C (TNC), pentraxin-related gene, rapidly induced by IL-1 beta (PTX3), and carboxypeptidase X (M14 family), member 1 (CPMX1). In chronic GO, but not in lymphedema, adipocyte-related immediate early genes known to be overexpressed in patients with active GO were upregulated but at a lower level than previously shown for the active phase. Genes of the Wnt pathway, such as secreted frizzled-related protein 1, 2, and 3, were up- and downregulated in both chronic GO and lymphedema. Parathyroid hormone-like hormone (PTHLH) was downregulated (p = 0.01) and apolipoprotein L domain containing 1 (APOLD1) was upregulated (p = 0.05) in both active and chronic GO.
Conclusions:
There are more differences than similarities between chronic ophthalmopathy and chronic lymphedema, but both conditions exhibit less inflammation and adipogenesis compared to the active phases. In lymphedema, fibrosis dominates. PTHLH, which can inhibit adipogenesis, is downregulated both in active and chronic ophthalmopathy, indicating the possibility of an increased risk of adipogenesis.
Introduction
Immediate early genes (IEGs) are overexpressed in active ophthalmopathy (17). One of these genes, cyclooxygenase 2 (COX-2), has a role in both inflammation and adipogenesis, and its expression increases with increasing disease activity (18).
Currently, there is no known preventive treatment for either disease, and the aim of this study was to better characterize the disease processes and investigate possible pathogenetic mechanisms shared between chronic GO and lymphedema.
Material and Methods
Collection of patient samples and isolation of RNA
All tissue samples were collected after informed consent with the approval of the Ethical Review Board of Lund University, Malmö/Lund, Sweden.
Subcutaneous adipose tissue was obtained from the arms of ten patients undergoing liposuction for chronic lymphedema. Control subcutaneous adipose tissue was collected from the unaffected arm of the same individuals. All patients were given general anesthetics, and before the start of liposuction, a biopsy was taken after a 1.5-cm incision on the medial aspect of the elbow of the normal and lymphedematous arm without the administration of local analgesia. Clinical characteristics are shown in Table 1A.
Used only for real-time polymerase chain reaction.
Intraorbital adipose/connective tissue was collected from ten female patients with chronic GO who underwent restorative surgery of the upper eyelid with opening of the orbital septum. The patients were operated 3–12 years after the diagnosis of ophthalmopathy, and the activity of their ophthalmopathy had been unchanged for at least one year before the operation. None of the patients had been treated with steroids within a year before surgery. Clinical characteristics of the patients are shown in Table 1B. Eight of these patients were analyzed with microarray (Patients 1–8 in Table 1B). These eight patients, plus the two additional patients not included in the microarray (Patient 9 and 10 in Table 1B), were analyzed by real-time polymerase chain reaction (PCR). Control intraorbital tissue was collected, after opening of the orbital septum, from female individuals (age 52–92 years) without thyroid disease undergoing cosmetic surgery of the upper eyelid. Eight controls were used for the microarray. For the real-time PCR, six samples included in the microarray and four additional controls were used (two samples used for the microarray were replaced by new samples due to a lack of material). For certain real-time PCR experiments, we also used intraorbital adipose/connective tissue from ten patients with severe active GO who underwent lateral decompression surgery (Table 1C). These patients were not included in our earlier expression profiling study of active GO (17).
RNA was extracted with the RNeasy Midi Kit (QIAGEN, Stockholm, Sweden) according to the manufacturer's instructions. RNA samples of 0.7 μg were combined into four pools (lymphedematous arm, healthy arm, chronic GO, and healthy intraorbital fat) with ten individual samples in the arm pools and eight in the intraorbital pools. After pooling, we used the RNeasy MinElute Cleanup Kit (QIAGEN) to obtain RNA at a higher concentration and purity for subsequent microarray analysis. RNA quality and concentrations were measured using an Agilent 2100 Bioanalyzer and Nanodrop ND-1000, respectively.
Microarray
The SCIBLU Microarray Resource Center at Lund University performed the gene expression analyses. By following the instructions of the GeneChip® Expression 3′-Amplification Reagents One-cycle cDNA synthesis kit (Affymetrix, Santa Clara, CA), 2.5 μg of total RNA was processed to produce double-stranded cDNA. This was used as a template to generate biotin-targeted cRNA following the manufacturer's specifications. Fifteen micrograms of the biotin-labeled cRNA was fragmented into strands between 35 and 200 bases in length. Ten micrograms of this cDNA was hybridized onto the GeneChip Human Genome U133 Plus 2.0 overnight in the GeneChip Hybridisation Oven 6400 using standard procedures. The arrays were then washed and stained using a GeneChip Fluidics Station 450. Scanning was carried out with the GeneChip Scanner 3000, and image analysis was performed using GeneChip Operating Software.
All of the arrays had 3′/5′ ratios of the housekeeping genes (beta-actin and glyceraldehyde-3-phosphate dehydrogenase) of less than 3 and a percentage present call rate of 45% or greater, which indicated a good and uniform in vitro transcription. We used the AffyProbeMiner (19) to regroup the individual probes into consistent probe-sets and remap the probe-sets to the correct sets of mRNA transcripts for the HG-U133 Plus 2.0 array, which resulted in 23,798 probe-sets. The normalization methods used for analysis of microarray data can have a major impact on the results of differential expression analyses (20). Therefore, we used six different algorithms for normalization and summarization, which combine the multiple probe intensities for each probeset to produce an expression value. The six normalization algorithms used were the MAS5.0 (21), the dChip-PM/MM difference model (22,23), the d-Chip PM Only model (22,23), the Robust Multi-array Average (RMA) method (24,25), the GC-Content Robust Multi-Array Average (GC-RMA) method (26), and the Probe Logarithmic Intensity Error (PLIER) method (Affymetrix). We conducted filtering based on the MAS5.0 present/absent calls, which classified each probe-set as expressed above background (present call) or not (absent or marginal call). We only included probe-sets that had a present detection call in at least one group (27), which allowed 16,005 out of 23,798 probe-sets to be further analyzed. In addition, we performed probe-level analysis using the significance score (S-Score) algorithm, which incorporates mixed-effects modeling of probe-level microarray data to identify differentially expressed genes between different groups (28,29). In interclass comparisons, we only considered probe-sets with at least a twofold change in all six algorithms for normalization and with an S-Score algorithm p-value <0.05 as significantly differentially expressed.
Sets of the differentially expressed genes were analyzed using the Web-based Gene Set Analysis Toolkit (WebGestalt) (30).
Real-time PCR
Total RNA (0.3 μg) was reverse transcribed in a 20 μL reaction following the instructions for the QuantiTect Rev. Transcription Kit (QIAGEN). PCRs were performed using the ABI PRISM 7900HT sequence detection system (Applied Biosystems, Stockholm, Sweden) according to the manufacturer's specifications. The reactions contained cDNA equivalent to 10 ng in a reaction volume of 10 μL using the TaqMan Universal PCR Master Mix (Applied Biosystems) and the TaqMan Gene Expression Assays (Applied Biosystems) following the manufacturers' protocols. The TaqMan Gene Expression Assays were chosen using the UmapIt Microarray-to-TaqMan Assays Mapping Tool (
Statistical analysis
The statistical significance of the difference in expression among groups was calculated using the Mann-Whitney rank-sum test, the Wilcoxon signed ranks test and the Kruskall-Wallis one-way analysis of variance with the SPSS 17.0 software (SPSS, Chicago, IL).
Results
Gene expression in adipose/connective tissue from the lymphedematous and normal arm in patients with chronic arm lymphedema
We identified 35 upregulated and 5 downregulated genes with microarray in the lymphedematous arm compared to the healthy arm (Table 2A1, A2). Enrichment analysis of the differentially expressed genes between the two groups with the Fisher's exact test using WebGestalt revealed a number of statistically significant ontologies. By performing further pathway analysis and a search of the literature, we identified several categories of genes with potentially important roles in the pathophysiology of the disease. These categories included differentiation and development, cytoskeleton components, wound healing and fibrosis, muscle contraction, adhesion, extracellular matrix (ECM) structure and remodeling, fat metabolism, inflammation, and the Wnt pathway. At least one gene from each category was chosen for replication with real-time PCR. Many of the selected genes had functions in several categories. The selected genes included the following: ABCG1 (fat metabolism, inflammation), SFRP2 (differentiation and development and the Wnt pathway), CPMX1 (differentiation and development and adhesion), DSP (differentiation and development, cytoskeleton components, wound healing and fibrosis, adhesion, and the Wnt pathway), PTX3 (inflammation), ACTA2 (cytoskeleton components, wound healing and fibrosis, and muscle contraction), and TNC (differentiation and development, wound healing and fibrosis, adhesion, ECM structure and remodeling, inflammation, and the Wnt pathway). All genes, except DSP, were upregulated two to three times greater in the lymphedematous arm compared to the healthy arm with real-time PCR (p < 0.05) (Fig. 1).
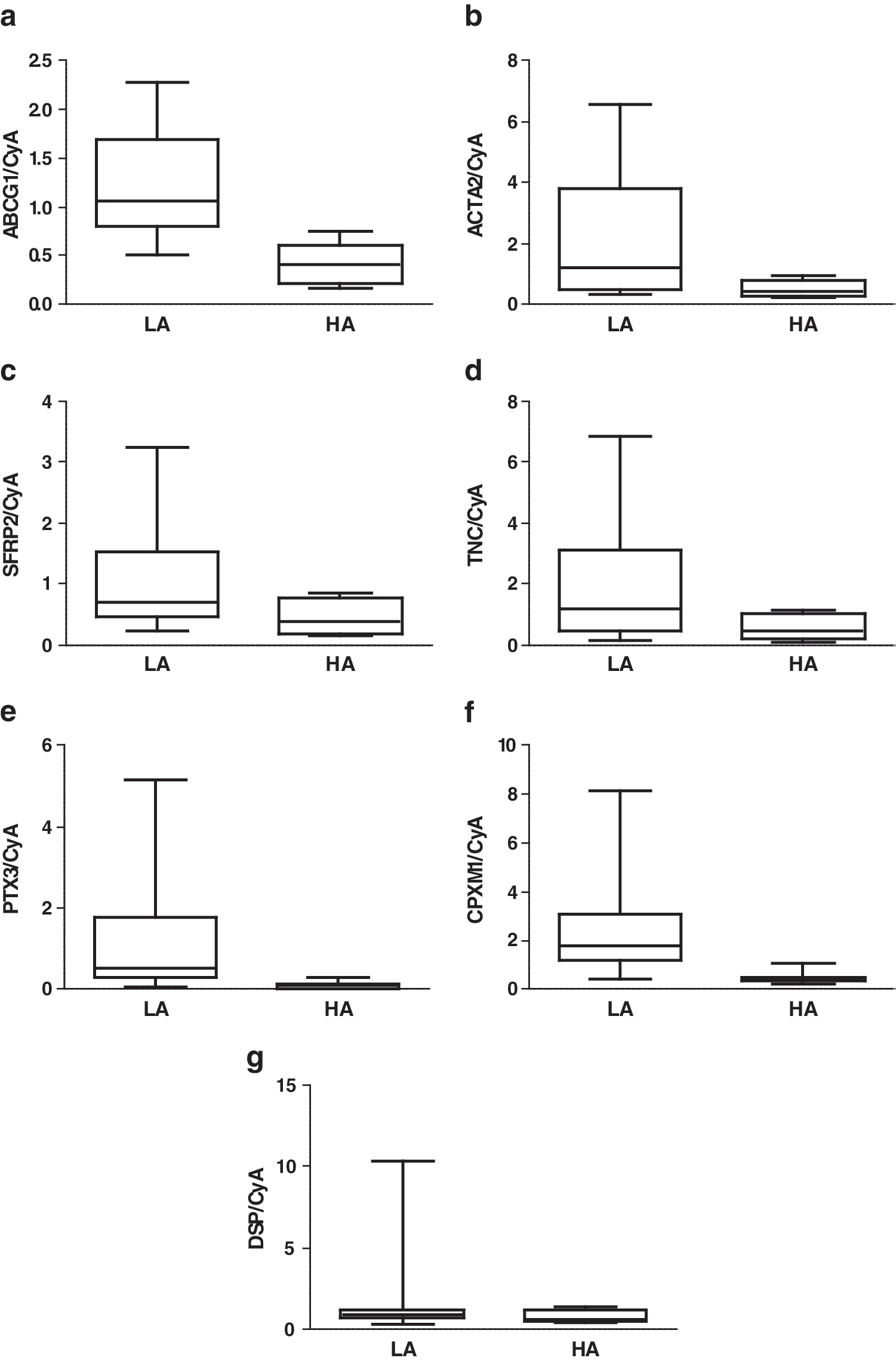
Expression of selected genes in subcutaneous adipose tissue of the LA and the HA of patients with chronic arm lymphedema (n = 10) as measured by real-time PCR and normalized to cyclophillin A. The same subjects were used as for the microarray. p-Values were calculated using the Wilcoxon Signed Ranks Test:
LA, lymphedema arm, HA, healthy arm; GO, Graves' ophthalmopathy; IOF, intraorbital fat.
Comparison of gene expression in chronic/active GO and chronic arm lymphedema
Adipocyte-related IEGs are known to be overexpressed in patients with active ophthalmopathy (17). In this microarray study of chronic GO, all but one of the eleven genes overexpressed in active GO were upregulated in the patient group compared to the controls at an average level of 1.4. This was in contrast to chronic arm lymphedema, where the IEGs were not upregulated (data not shown).
In our previous microarray of active GO (17), enrichment analysis revealed statistically significant upregulation of genes with functions in the inflammatory and immune response (unpublished data). This significant upregulation was not observed in chronic GO and chronic lymphedema, where only one gene in each condition (v-fos FBJ murine osteosarcoma viral oncogene homolog [FOS] in chronic GO and PTX3 in chronic lymphedema) was upregulated more than twofold out of the 31 upregulated genes in active GO (Table 3). The upregulation of nuclear receptor subfamily 4, group A, member 2 (NR4A2), interleukin 6 (IL6), interleukin 8 (IL8), and PTX3 in active GO was also confirmed with real-time PCR.
Data are presented as expression ratio cases/controls (fold change). Upregulation more than twofold is marked in bold.
NA, not analyzed.
Several genes involved in the Wnt pathway were identified in the microarray as upregulated, but with different expression patterns, in both the lymphedematous arm compared to the healthy arm (SFRP2, DSP, FHL2, and TNC) and in chronic GO compared to healthy intraorbital adipose tissue (RSPO2 and AXUD1). The overexpression of SFRP2 in the lymphedematous compared to the healthy arm was confirmed by real-time PCR (Fig. 1c). Another secreted frizzed related protein (SFRP), SFRP1, was previously found to be upregulated in active GO in a different study (31). Therefore, we studied the expression of three SFRPs in the active and chronic phases of ophthalmopathy and in controls with real-time PCR. In our study, SFRP3 was downregulated in both the active and chronic phase compared to controls (p = 0.01) (Fig. 2c). SFRP1 and SFRP2 showed no upregulation in active or chronic GO compared to controls (Fig. 2a, b).
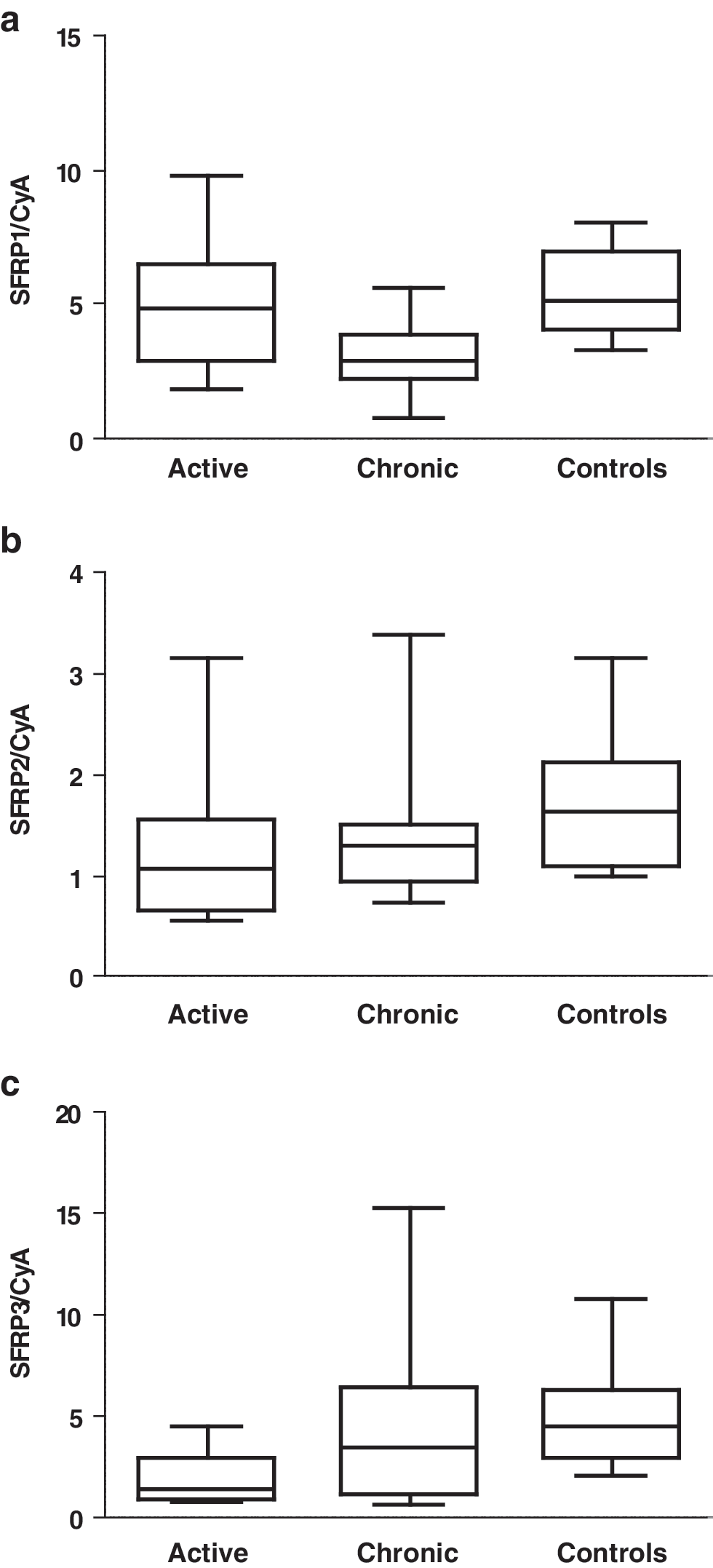
Expression of secreted frizzled-related proteins (SFRPs) in intraorbital adipose/connective tissue of patients with active Graves' ophthalmopathy (n = 10), patients with chronic Graves' ophthalmopathy (n = 10), and thyroid-healthy controls (n = 10) as measured by real-time PCR and normalized to cyclophillin A. RNA from the same patients as for the microarray and from two new patients was used. Two of the controls used for the microarray had to be replaced by new controls due to a lack of material, and two additional controls were used. p-Values were calculated using the Kruskal-Wallis Test:
Gene expression in intraorbital adipose/connective tissue from GO patients in the active or chronic phase and thyroid-healthy controls
The microarray analyses identified 14 upregulated genes and 22 downregulated genes in chronic GO tissue compared to the healthy intraorbital tissue (Table 2B1, B2). Enrichment analysis of the differentially expressed genes between the two groups with the Fisher's exact test using WebGestalt gave statistically significant ontologies for the downregulated genes, but none for the upregulated genes. Two of the upregulated genes and several downregulated genes were up- and downregulated, respectively, in our earlier microarray study of active ophthalmopathy (17), but have not been confirmed with real-time PCR. Therefore, we chose the two upregulated genes, FOSB and APOLD1, and one of the downregulated genes, PTHLH, for analysis with real-time PCR. With real-time PCR we confirmed the upregulation of APOLD1 (p = 0.05) and the downregulation of PTHLH (p = 0.01) in both chronic and active phases. The upregulation of FOSB was confirmed for the active but not for the chronic phase (Fig. 3).
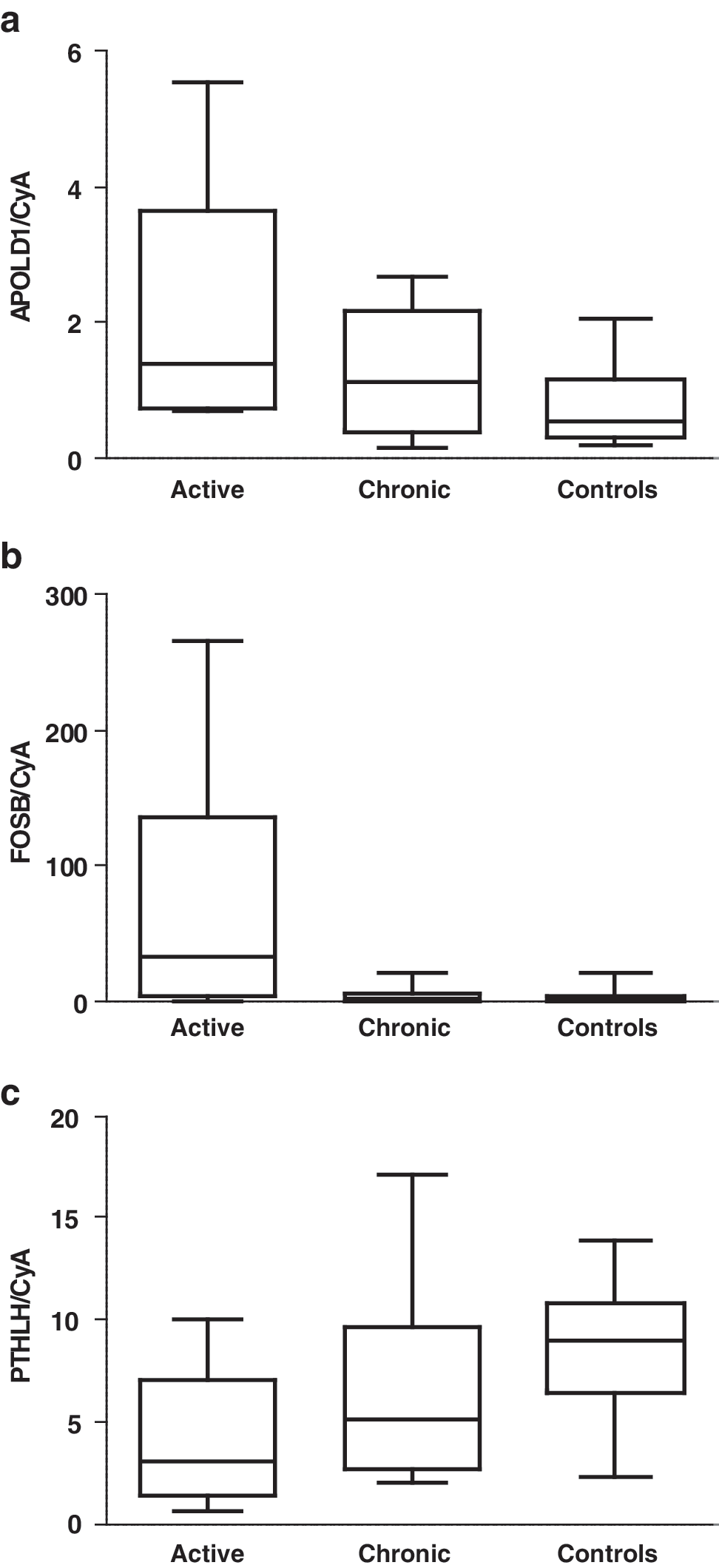
Expression of selected genes in intraorbital adipose/connective tissue of patients with active (n = 10) and chronic (n = 10) Graves' ophthalmopathy and thyroid-healthy controls (n = 10) as measured by real-time PCR and normalized to cyclophillin A. RNA from the same patients as for the microarray and from two new patients was used. Two of the controls used for the microarray had to be replaced by new controls due to a lack of material, and two additional controls were used. p-Values were calculated using the Kruskal-Wallis Test:
Discussion
This study is currently the first large-scale expression profiling of chronic human arm lymphedema and chronic GO. In arm lymphedema, there is a distinct gene expression pattern that is different from active and chronic ophthalmopathy. This expression pattern involves upregulation of genes with roles in wound healing, formation of ECM, and the Wnt system, but not upregulation of genes with functions in early active adipogenesis, as the IEGs. This finding is in contrast to chronic/active ophthalmopathy, where adipogenesis genes are upregulated and follow the disease activity. In addition, we found two genes, PTHLH and APOLD1, that were down- and upregulated, respectively, in both active and chronic GO. We also demonstrated that some of the Wnt modulators were up- or downregulated in both lymphedema and GO, but with distinct expression patterns.
When comparing the results from chronic GO to our earlier microarray study of active ophthalmopathy (17), there was less activity in the chronic phase, that is, a lower number of up- or down-regulated genes. In active ophthalmopathy, 167 genes were upregulated and 23 downregulated in the patient group compared to the controls (17). In the current study of chronic ophthalmopathy, only 14 genes were upregulated and 22 downregulated, and a similar situation was observed in lymphedema. Gene expression profiling of acute lymphedema has not been performed in humans but has been performed in a mouse model (11). In the animal study, 429 genes were upregulated and 183 were downregulated, whereas 35 genes were upregulated and 5 were downregulated in our human study. Four genes were upregulated in both studies: ACTA2; TNC; lectin, galactoside-binding, soluble, 7 (LGALS); and transgelin (TAGLN).
Another clear difference between the acute and chronic phases of both conditions is the absence of pronounced inflammation. In the microarray study of active ophthalmopathy, many genes involved in inflammation and immune response were upregulated. Upregulation of acute inflammatory genes was not observed in chronic GO and some genes involved in inflammation, such as the chemokines CCL13 and CCL18 or dipeptidyl-peptidase 4 (DPP4), were even downregulated in the chronic phase. In the mouse study of acute lymphedema, a large number of genes involved in acute inflammation was upregulated (11), but in the current study, only three of the upregulated genes, ABCG1 (32), PTX3 (33), and TNC (34), have previously been implicated in inflammation.
The two previous gene expression profiling studies on active ophthalmopathy demonstrated that orbital adipogenesis was enhanced both in the early proliferation phase and in the terminal differentiation phase (17,31). The major finding of our earlier study of active ophthalmopathy was the overexpression of adipocyte-related IEGs (17), which are known to initiate the mitotic clonal expansion phase during adipogenesis in a mouse cell line (35). In the current microarray study of chronic GO, we found higher expression of most of these genes in the patient group compared to the controls but at a lower level than what has been shown for the active phase. Kumar et al. (31) observed upregulation of SFRP1 and suggested that modulation of the Wnt system may have a role in orbital adipogenesis. We therefore searched for genes on our arrays with known functions in the Wnt system. Thus, we studied the expression of three SFRPs in patients with active and chronic GO with real-time PCR, but we could not confirm the upregulation of SFRP1 observed by Kumar. One explanation may be the use of different controls. Kumar et al. used orbital tissue from autopsied patients. However, we observed downregulation of SFRP3 in patients with both active and chronic GO. In other cell systems, SFRP3 was downregulated in response to stimulation by growth factors or mechanical trauma (36,37). In patients with GO, the expression of several growth factors was enhanced (data not shown), and the increased intraorbital pressure causes mechanical stress.
Patients with chronic arm lymphedema following breast cancer treatment were proven to have excess adipose tissue by volume-rendered computed tomography and dual energy X-ray absorptiometry (16). Surprisingly, we did not observe upregulation of any key genes involved in active adipogenesis in lymphedema, however. These results suggest that in the late stages of chronic lymphedema, active adipogenesis diminishes, and the excess adipose tissue found during operation may be a consequence of pronounced adipogenesis at earlier stages.
Although the patients underwent operation for lymphedema 3–24 years after the breast cancer operation, a whole panel of genes involved in wound healing was upregulated. It is well known from other fibrotic disorders that chronic inflammation can cause deregulation of wound healing and fibrosis (38). The myofibroblast is a key cell involved in the process of fibrosis. After complete healing, myofibroblasts normally undergo apoptosis. In chronic lymphedema, this mechanism seems to fail, which leads to the persistence of myofibroblasts with deposition of ECM and scarring. The fully differentiated myofibroblast is characterized by expression of markers such as ACTA2 and desmin (DES) [reviewed in ref. (38)], which were upregulated in chronic lymphedema in this study. A number of other genes with functions in wound healing and fibrosis were also upregulated in our study: LGALS7, TAGLN, TNC, cartilage oligomeric matrix protein (COMP), desmoplakin (DSP), desmocollin 1 (DSC1), four and a half LIM domains 2 (FHL2), periostin, osteoblast specific factor (POSTN) and stratifin (SFN). Because excessive production of ECM is another hallmark of fibrosis, the upregulation of a group of genes involved in ECM structure and remodeling, POSTN, TNC, COMP, and SPARC related modular calcium binding 2 (SMOC2) further supports the hypothesis that fibrosis is the key process in chronic lymphedema. Upregulation of genes involved in fibrosis was not observed in patients with chronic GO.
To find genes not varying with disease activity, we looked for genes that were up- or downregulated both in active and chronic phases of GO. An interesting observation is the downregulation of PTHLH in both active and chronic GO and not in lymphedema. PTHLH is a multifunctional protein that may have evolved to regulate local tissue functions in contrast to parathyroid hormone, with systemic hormonal effects. One function of PTHLH is the inhibition of adipogenesis (39,40), and if downregulated, it may promote adipogenesis. Whether this observation is a consequence of the disease process or is inherited needs be further investigated.
To summarize, in this study of chronic arm lymphedema and chronic GO, we observed less inflammation and adipogenesis compared to what has been shown for the acute phases of both conditions. The main difference between GO and lymphedema seems to be the profound fibrosis in lymphedema not observed in chronic GO and the absence of active adipogenesis in chronic lymphedema. Fibrosis is often irreversible, and future therapeutic strategies should focus on prevention. We observed downregulation of PTHLH, a gene with known functions in adipogenesis, in both active and chronic GO, which indicates the possibility of an inherited risk of adipogenesis. Because only one-third of Graves' patients and breast cancer operated patients develop ophthalmopathy and lymphedema, respectively, it will be important to investigate whether some of the above described genes can be used as markers and treatment targets for patients at risk for developing these diseases.
Footnotes
Acknowledgments
We are grateful to Gertrud Ahlqvist for handling the tissue biopsies. We acknowledge help with the microarray laboratory work and analysis from the SCIBLU Microarray Resource Centre at Lund University. This study was supported by grants from the Research Funds of Skåne University Hospital, Skåne Research Foundation, and The Faculty of Medicine at Lund University.
Disclosure Statement
The authors declare that no competing financial interests exist.