
Abstract
Background:
Phosphatase and tensin homolog deleted on chromosome ten (PTEN)-hamartoma tumor syndrome (PHTS) is a complex disorder caused by germline inactivating mutations of the PTEN tumor suppressor gene. PHTS includes Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRRS), and Proteus-like syndromes. Affected individuals develop both benign and malignant tumors in a variety of tissues, including the thyroid. This study is to better characterize and describe the thyroid pathology within the different entities of this syndrome, and examine whether there is an association between specific thyroid findings and different PTEN mutations.
Methods:
Twenty patients with known PTEN mutations, and/or clinical diagnosis of PHTS, and thyroid pathology were identified: 14 with CS and 6 with BRRS.
Results:
Thyroid pathology findings were as follows: multiple adenomatous nodules in a background of lymphocytic thyroiditis (LT) in 75%, papillary carcinoma in 60%, LT alone in 55%, follicular carcinoma in 45%, C-cell hyperplasia in 55%, and follicular adenomas in 25%. Within the papillary carcinoma group, there were 6 microcarcinomas, 5 follicular variants, and 1 classical type.
Conclusions:
There were no morphologic differences between the thyroid findings in CS and BRRS. Also, there was no correlation between specific PTEN germline mutations (exons 5, 6, and 8) and pathologic findings. Distinctive and characteristic findings in PHTS include multiple unique adenomatous nodules in a background of LT, and C-cell hyperplasia; it is vital that pathologists recognize the classical histologic features of this syndrome to alert clinicians to the possibility of this syndrome in their patients.
Introduction
Materials and Methods
All thyroid lesions from patients with known PTEN mutations and/or clinical diagnosis of CS or BRRS were retrieved from the Departments of Pathology of Brigham and Women's Hospital, Children's Hospital Boston, Boston, MA, and Hopitaux Universitaires de Geneve, Switzerland. Specimen photographs, histopathology, and clinical and molecular findings were reviewed. Cases were classified according to the World Health Organization (WHO) classification of thyroid tumors (2). In addition, several studies addressing follicular lesions were reviewed (10 –19). Calcitonin immunostaining was performed using a prediluted polyclonal antibody (Cell Marque Corporation, Hot Springs, AZ). C-cell mapping was performed and population density was estimated using the following criteria, as set forth in previous studies (9,20,21): immunostained sections were scanned at low magnification (× 100) to detect clusters of C-cells, which were then enumerated at high magnification (× 400). When a cluster of at least 50 C-cells in one low power field (× 100) was identified, a diagnosis of C-cell hyperplasia (CCH) was made. Germline mutations of PTEN were analyzed by bidirectional DNA sequence analysis of the 9 exons of the PTEN gene and their respective splice sites (22).
To illustrate the clinical complexity of this syndrome, we have summarized the clinicopathologic findings in these patients in Tables 1 and 2.
Age at diagnosis of CS/BRRS.
BRRS, Bannayan-Riley-Ruvalcaba syndrome; Ca, carcinoma; CS, Cowden syndrome; ER, estrogen receptor; F, female; GI, gastrointestinal; M, male; MGM, maternal grandmother; MGF, maternal grandfather; N/A, not available; PGF, paternal grandfather; PGM, paternal grandmother; PR, progesterone receptor; PTEN, phosphatase and tensin homolog deleted on chromosome ten.
Defined as >50 cells/100× field.
FC, follicular carcinoma; PTC, papillary thyroid carcinoma; PTC (micro), papillary thyroid microcarcinoma; MI, minimally invasive; LT, lymphocytic thyroiditis; MAN, multiple adenomatous nodules.
Results
Pathologic findings
Of the 20 patients with PHTS (CS [n = 14]; BRRS [n = 6]), 14 were female and 6 were male. The majority (11/20, 55%) had a family history of PHTS, and the remainder (9/20, 45%) were classified as simplex (no apparent family history); all patients fulfilled the diagnostic criteria for PHTS. The mean age at diagnosis of PHTS was 33.7 years (9–76 years). When subclassified into the two syndromes, the mean age at diagnosis of BRRS was 15 years (9–23 years) and 41.7 years (13–76 years) for CS. The mean age at diagnosis of a thyroid lesion was 33.3 years (range 7–73 years); 15 years (7–24 years) for BRRS, and 41.1 years (22–73 years) for CS. Thus, identification of a thyroid lesion and a diagnosis of PHTS were often concurrent (Tables 1 and 2). Further inspection revealed that in 15 patients (cases 2–7, 11–14, and 16–20) the thyroid diagnosis was made at the same time as the PHTS diagnosis. In three patients, the PHTS diagnosis preceded the thyroid diagnosis (case 1 by 1 year, case 8 by 10 years, and case 10 by 2 years), and in two patients, the thyroid pathology diagnosis preceded the ascertainment of PHTS (case 9 by 8 years, and case 15 by 13 years). For some patients, thyroid pathology was diagnosed during evaluation and screening for PHTS, and for others, the PHTS diagnosis and work-up, including PTEN mutation analysis, was triggered by pathologic findings.
Seventeen patients underwent a total thyroidectomy, and the remaining three opted for lobectomy. Tumors were classified according to the World Health Organization classification system (2), and all thyroids had multiple pathologic findings (Table 2). The most common diagnosis was MAN in 75% (n = 15; Figs. 1 –3), followed by PTC in 60% (n = 12; 4 multifocal follicular variants, 1 follicular variant, 1 classic type, and 6 microcarcinomas), lymphocytic thyroiditis (LT) in 55% (n = 11; Fig. 2b), CCH in 55% (n = 11, Fig. 4), FC in 45% (n = 9), nodular hyperplasia in 25% (n = 5), and multiple and solitary follicular adenomas in 25% (FA, n = 5).
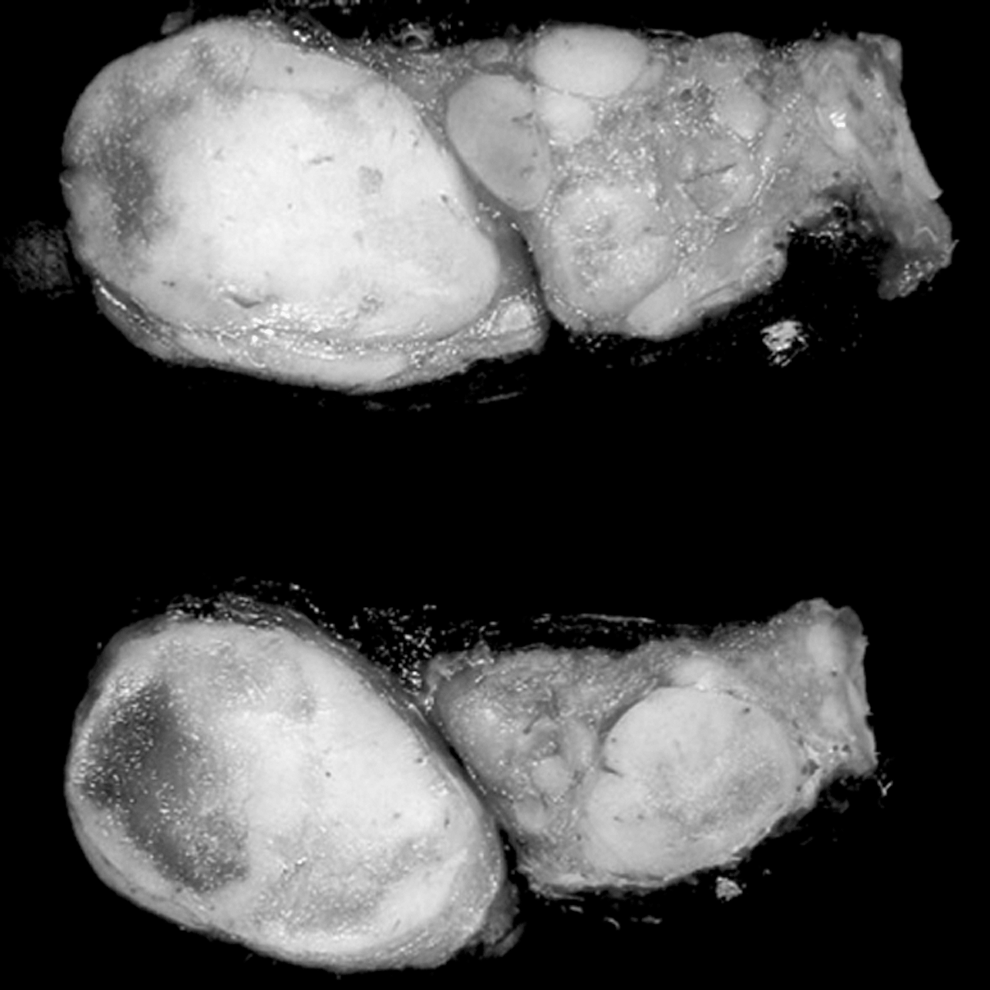
Thyroid cut surface. Multiple adenomatous nodules (MAN), with the characteristic macroscopic appearance showing well-circumscribed, solid, tan nodules. Close-up view of the cut surface.
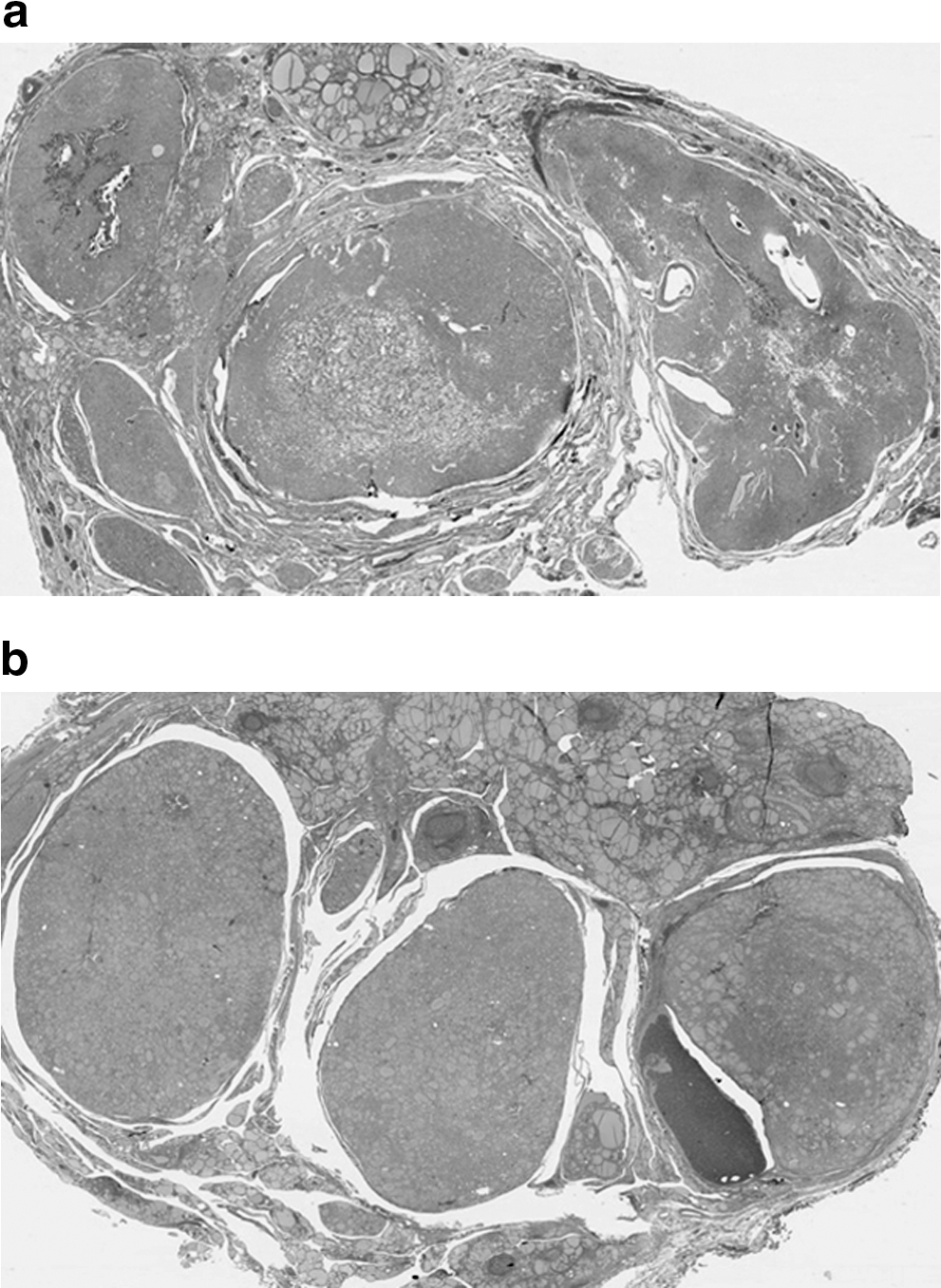
Scanning view of thyroid with MAN. MAN with little intervening normal thyroid
MAN. Nodules are well-circumscribed but unencapsulated. They have a solid and cellular appearance and are separated by scattered residual follicles (H&E; 100 ×).

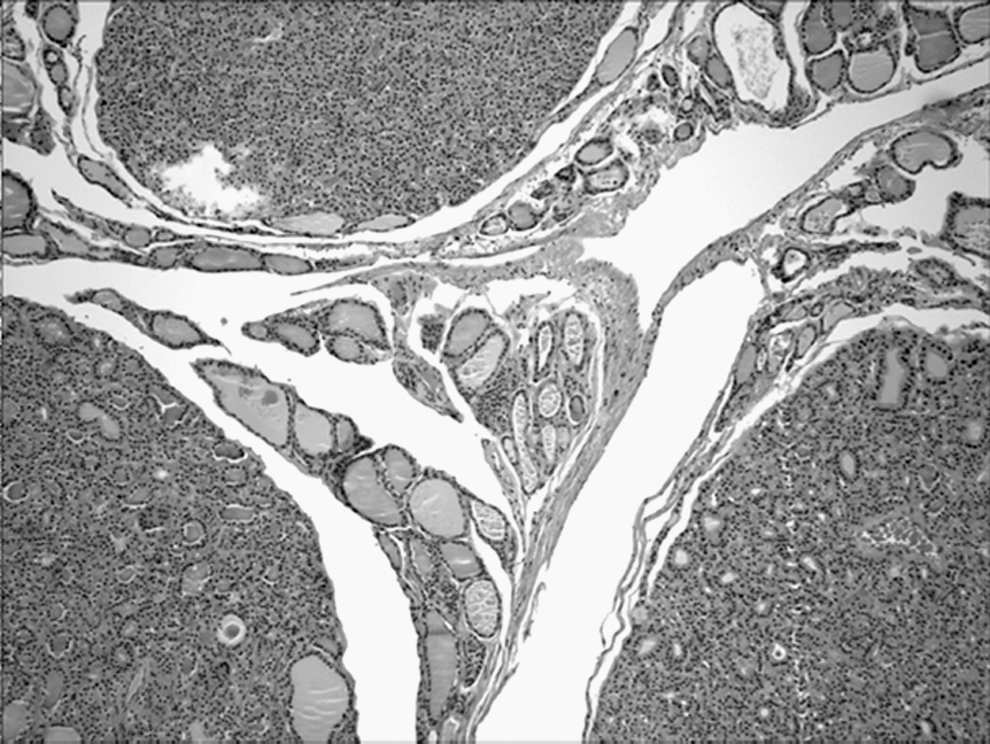
C-cell hyperplasia
MAN in a background of LT was the most frequent histologic finding; MAN were diagnosed in 15 specimens, which represented 50% (3/6 cases) of BRRS patients and 86% (12/14 cases) of CS patients in this series. The size of the adenomatous nodules ranged from 0.3 to 8.5 cm in greatest dimension, and the number of nodules per gland ranged from over 20 to more than 100 (Table 2). Grossly, these nodules were numerous, firm, white/tan, and homogenous, without gelatinous colloid or prominent secondary change, distinguishing them from the more common nodular hyperplasia. Microscopically, these adenomatous nodules had a solid architecture with a cellular composition, frequently lacking colloid, and without prominent cystic degeneration or heterogeneity among nodules. The majority of nodules were well-circumscribed and unencapsulated, though a few had a discontinuous rim of thin fibrous tissue simulating a capsule. Adjacent normal thyroid tissue was scanty, compressed, and consisted of a thin rim of occasional follicles in a background of LT. There were no histologic differences between the adenomatous nodules in CS versus BRRS.
CCH, as shown by calcitonin immunostaining (Fig. 4), was also a frequent finding; it was seen in 55% (11/18) of all patients. Of these, five were in BRRS patients (5/6, 83%) and six in CS patients (6/14, 43%). Thus, CCH can be seen in any PHTS patient. Although CCH is not as well defined in the pediatric age group (23,24), it appears to be relatively more common in BRRS patients.
Thyroid carcinoma was identified in the majority of patients in our study (15/20, 75%) (Table 2). For FC specifically, the average age at diagnosis in PHTS was 22.1 years (7–53 years); 32 years (18–53 years) in CS patients, and 14 years (7–24 years) in BRRS. Of the patients with CS, 35% (5/14 cases) had FC, only one of which was multicentric. In addition, we found FC in four patients with BRRS (4/6 cases; 67%), indicating that FC was frequent in the pediatric BRRS population. None of the malignancies were diagnosed preoperatively; all carcinomas were diagnosed by careful pathological examination.
Unexpectedly, we also identified PTCs (12/20 cases, 60%), which have rarely been described in these patients; PTCs occurred in patients with CS (9/14 cases, 64%) and BRRS (3/6 cases, 50%). Six were microcarcinomas (cases 1, 3, 10, 14, 16, and 19) ranging in size from 0.15 to 0.5 cm, four were multifocal follicular variant of PTC (cases 2, 6, 9, and 15), one case was a classic type PTC (case 5), and one was a follicular variant of PTC (case 19) (the thyroid pathology findings and size for all malignant tumors is included on Table 2). The mean age at diagnosis of PTC in PHTS was 32.4 years (11–73 years), 31.6 years in CS (18–73 years), and 17.5 years in BRRS (11–24 years).
In our series, as in the literature, the most commonly reported extrathyroidal malignancy was ductal breast carcinoma (5/20 cases), all of which occurred in CS patients. Two tumors were invasive ductal carcinoma (cases 3 and 9), two were ductal carcinoma in situ (cases 2 and 4), and additional information was not available in one case (case 14). The mean age at diagnosis of patients with ductal breast carcinoma was 48.9 years (33–69 years). One extrathyroidal malignancy was reported in our BRRS patients (case 15, renal cell carcinoma at age 11). No uterine malignancy was documented in our series.
Molecular genetics findings
Analysis of germline PTEN mutations was available for review in 13 patients; no mutation was identified in one patient, and six patients (although they fulfilled the diagnostic criteria for PHTS clinically) either declined mutation testing or requested that their results be withheld. The results of the mutation analyses are summarized in Table 3.
→ , leading to; AA, amino acid; FS, frame shift; PT, premature termination.
The majority of mutations occurred in exon 5 (7/13, 54%), followed by exon 6 (3/13, 23%) and exon 8 (3/13, 23%), confirming, as previously reported, that the most frequent site of germline mutations in PHTS is exon 5. Mutations identified in exon 5 were as follows: five missense mutations, one nonsense mutation, and one deletion that produced a frame shift leading to a premature termination. Exon 6 showed deletions and/or insertions, and exon 8 revealed two nonsense mutations, and a duplication that produced a frame shift leading to a premature termination.
Germline PTEN mutations and their associated pathologic findings are summarized as follows: exon 5 mutations (patients 1, 2, 8, 15, and 18–20) had FC, MAN, PTC, nodular hyperplasia (NH), and FA; exon 6 mutations (patients 5, 10, and 11) had FC, PTC, and FA; and exon 8 mutations (patients 7, 16, and 17) had FC, MAN, NH, and FA. Thus, we found no correlation between specific germline mutations and thyroid pathologic findings.
Discussion
CS and BRRS (Tables 4 and 5) are the major entities comprising the PHTS, which is caused by germline mutations of the PTEN gene and inherited in an autosomal dominant fashion (4).
Diagnostic criteria refer to the current operational diagnostic criteria for CS, which are available online at the National Comprehensive Cancer Network (
LDD, Lhermitte-Duclos disease.
BRRS generally presents in childhood with delayed motor and intellectual development, and other described features include thyroid adenomas, LT/Hashimoto's thyroiditis, lymphatic/vascular malformations, high arched palate, scoliosis, seizures, lipid storage myopathy, and joint hyperextensibility (5,19,43).
PTEN is a tumor suppressor gene located on 10q23.3 (1). The function of PTEN is not entirely understood, but it is a major phosphatase for phosphoinositide-3,4,5-triphosphate. By downregulating the levels of phosphoinositide-3,4,5-triphosphate, PTEN produces an inhibitory (tumor suppressor) effect on the PI3P/Akt pathway, an important carcinogenesis pathway. It is proposed that PTEN has important activity both in the cytoplasm and nucleus. Nuclear PTEN might be required for cell cycle arrest by downregulating cyclin D1 and preventing phosphorylation of mitogen-activated protein kinase pathway, whereas cytoplasmic PTEN seems to be required for apoptosis by downregulating the phosphorylation of AKT and upregulating p27. Therefore, loss of PTEN function results in escape from programmed cell death and G1 arrest in the cell cycle. The gene is composed of 9 exons, with a 1209-bp coding sequence, which is predicted to encode a 403-amino acid protein. Mutations are dispersed throughout the 9 exons of the PTEN gene, with ∼40% of mutations located in exon 5, which represents 20% of the coding sequence and is known as the mutational “hot spot” in PHTS. Very few germline mutations have been reported in exon 1, and none in exon 9 (25). Seventy-five percent of the germline mutations result in truncated protein, lack of protein, or dysfunctional protein (26 –31).
In contrast to medullary thyroid carcinoma (MTC), where up to a quarter is associated with a familial syndrome, only 5% of non-MTCs are associated with a heritable condition. These familial non-MTC can be divided into two distinct groups (8); those occurring in tumor syndromes where thyroid involvement is a minor component and those occurring in syndromes where non-MTCs are the predominant tumor encountered. The first group includes familial adenomatous polyposis, Carney complex type 1, Werner syndrome, and the focus of this study, PHTS. The second group of familial non-MTC syndromes, in which non-MTCs are the major finding, includes the following rare entities; pure familial PTC (fPTC), with or without oxyphilia, fPTC with papillary renal cell carcinoma, and fPTC with multinodular goiter (2,4,6,8).
Although PTEN mutations define this syndrome, they are rare in sporadic thyroid carcinomas (32). In CS, germline intragenic PTEN mutations have been found in up to 81% of classically affected patients and gene deletions have been described in very rare cases (5,30). In BRRS, intragenic mutations are seen in 60% of patients (5,30). In our series, the majority of mutations occurred in exon 5 (7/13 cases, 54%), followed by exon 6 (3/13 cases, 23%) and exon 8 (3/13 cases, 23%), confirming that the most frequent site of germline mutations in PHTS is exon 5. Case 3 (CS) had a negative mutation test, which is expected to occur in ∼10%–15% of individuals who meet diagnostic criteria for CS (25). However, the specific germline mutation does not correlate with specific thyroid pathology (Tables 2 and 3), including CCH.
Studies have failed to demonstrate a consistent genotype–phenotype relationship in PHTS (22,25,30,33,34). The earliest phenotypic features associated with PTEN mutations are macrocephaly and mucocutaneous lesions. Hamartomas, including gastrointestinal polyps, are often seen, and malignancies develop over time (33). BRRS often presents during childhood, and as of yet, an international consensus on the criteria for diagnosis has not been reached. Marsh et al. (22) defined the criteria as least three of the following four features: macrocephaly, lipomatosis, hemangiomas, and speckled penis. Parisi et al. (35) defined BRRS as at least two of three features, including macrocephaly, hamartomas (at least one lipoma, hemangioma, or intestinal polyp), and penile macules. Of note, it has recently been shown that the hemangiomas in BRRS are intramuscular hamartomatous proliferations of abnormal blood vessels and fibroadipose tissue (36). Other features described in BRRS include thyroid adenomas, Hashimoto thyroiditis, and lymphatic malformations, among others (30,37) (Table 5). In contrast, CS has well-established diagnostic criteria (Table 4). Careful phenotype analysis will provide support for the assertion that BRRS and CS are actually one condition, presenting at different stages (33). This study helps to support this observation by the lack of genotype–phenotype correlation in the thyroid gland.
More than 90% of individuals affected with CS manifest a phenotype by age 20 (6,38). By the end of their third decade, nearly all (99%) develop the pathognomonic mucocutaneous lesions, although any feature may be present, including thyroid lesions (Table 4). The spectrum of disorders related to PHTS has continued to evolve since 1995, when diagnostic criteria for CS were introduced. The current diagnostic criteria for CS consider FC (present in 10%–15%) a major criterion, whereas multinodular goiter and follicular adenomas are minor criteria (present in 50%–67%) (5,38,39). Our histologic findings in this study suggest that the multinodular goiter seen in PHTS patients is commonly due to characteristic, distinct MAN, rather than the adenomatous changes of old age. Nodular hyperplasia, a common thyroid disease, has a heterogenous, hyperemic gross appearance with nodules containing gelatinous colloid, and frequent secondary changes such as calcification and cystic degeneration. Histologically, they are unencapsulated, heterogenous, and composed of large, irregular, dilated follicles with hyperplastic epithelium and abundant colloid; only occasional nodules are densely cellular. In contrast, the adenomatous nodules in PHTS are firm, yellow-tan, lack gelatinous colloid and do not exhibit secondary changes. Histologically, the adenomatous nodules are solid, cellular, and composed of small follicles lacking abundant colloid. Some nodules may have a thin, discontinuous rim of fibrous tissue simulating a capsule.
In our experience, the MAN seen in PHTS are bilateral, more numerous, and distinct from age-related nodules, with only a scant atrophic rim of normal thyroid follicles. The number of these nodules far exceeds those seen in old age, or in any other inherited syndrome; in this series we report MAN in 75% of PHTS patients (50% of those with BRRS patients and 86% of those with CS). Nodule size (0.3–8.5 cm) and number per thyroid (20–>100) varied widely, but morphologic differences between the lesions of CS and BRRS were not identified. We are of the opinion that the International Cowden's Consortium CD Diagnosis Criteria/National Comprehensive Cancer Network consider adding MAN as a major criterion for the diagnosis of CS and BRRS (5,34,38 –41).
FC is an important feature of both CS and BRRS. It has been reported that these tumors are frequently multicentric, and are believed to arise from pre-existing follicular adenomas (2,3) We too have found that the majority of carcinomas arise in a background of MAN, though in our series only one FC (case 13) had a multicentric pattern. Management for CS patients has focused on cancer screening, and surveillance recommendations include annual thyroid ultrasounds after the age of 18 (22). If a thyroid nodule measures ≥1 cm, it is recommended that it be evaluated with fine needle aspiration, followed by standard management with surgery and adjunctive therapy if indicated (42,43). In contrast to other familial syndromes (44), prophylactic thyroidectomy in PHTS is not recommended. A cancer risk in BRRS patients has previously been suspected, though unknown (25,33,37); we now report a high frequency of FC (4/6 cases, 67%) in our series, with a mean age at diagnosis of 14 years. Given these findings, as well as the identification of renal cell carcinoma in a young BRRS patient (case 15), we believe that BRRS patients should be followed with similarly rigorous surveillance.
The majority of thyroid lesions occurring in PHTS are of follicular origin; papillary carcinoma has been only rarely associated with this entity (3,7,9). In our study, we identified PTC in 60% of patients (12/20 cases). However, given the prevalence of PTC in the general adult population (25%–34%) (45,46), it is difficult to conclude from these data that PTC is characteristic of PHTS. We do not endorse the addition of PTC as a diagnostic criterion for either CS or BRRS.
MTC is not considered part of the spectrum of PHTS; however, studies have identified CCH in individuals with this syndrome (3,9). C-cell proliferations may be either neoplastic or physiologic; within the neoplastic spectrum, calcitonin producing C-cells have been categorized as a precursor of familial MTC (21,44,47). Physiologic CCH is a reactive process associated with hypercalcemia, hypergastrinemia, goitrous hypothyroidism, Hashimoto thyroiditis, and attributed to peritumoral effect (adjacent to large papillary or follicular neoplasms, and non-Hodgkin lymphoma of the thyroid) (20,23,24). Interestingly, CCH was identified in 11 patients in this series: 5 with BRRS and 6 with CS. The pattern and degree of hyperplasia varied from focal and patchy to diffuse, and was associated with a variety of lesions (Table 2). These findings were most prevalent in BRRS patients, which could represent an age-related phenomenon (9,23,24,40). However, the high number (>>50 per 100 × field) of C-cells, often exceeding the number of follicular cells, more plausibly points to an abnormality of C-cells in PHTS, although the criteria for CCH are not well defined for the pediatric age group. CCH was not associated with age in our study. We recommend that pathologists include PHTS in the differential diagnosis of CCH of the thyroid.
The diagnosis of a thyroid lesion preceded the diagnosis of PHTS by many years in some patients in this study, whereas in others, a work-up for PHTS (including PTEN mutation analysis) was triggered by pathologic findings in the thyroid (specifically, MAN). We propose an algorithmic approach for the pathologist adressing unusual thyroid pathology in a young patient (Fig. 5). MAN, associated with other thyroid lesions, CCH, and/or FC in young patients should alert the pathologist to the possibility of an inherited tumor syndrome.
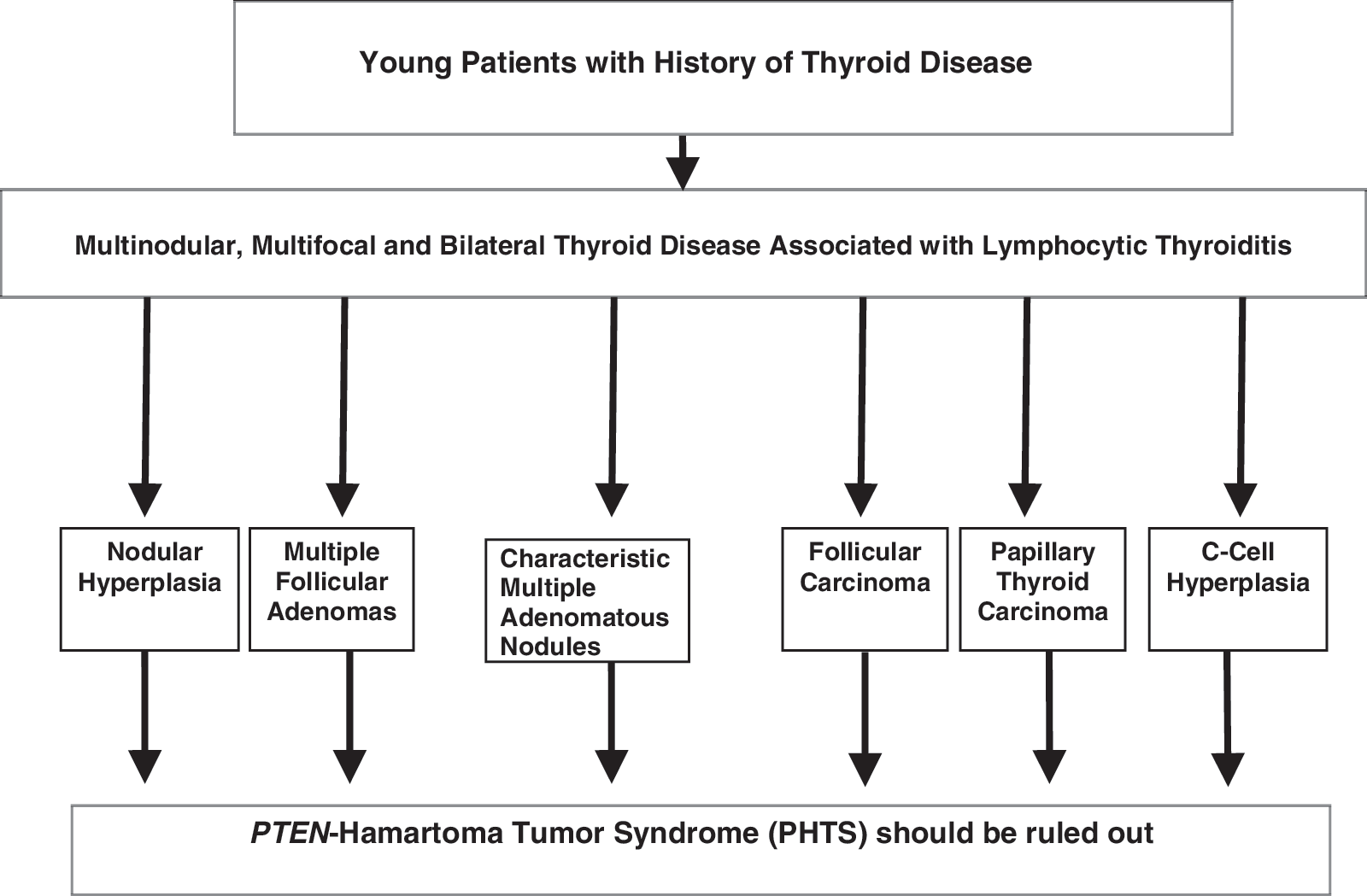
Algorithmic approach to unusual thyroid pathology in young patients.
In conclusion, characteristic multicentric, bilateral, benign, and malignant thyroid lesions are observed in PHTS. Morphologic differences between thyroid lesions in CS and BRRS were not observed, although CCH was more frequent in the younger BRRS patients. There was no correlation between specific germline mutations and pathologic findings identified. This supports the belief that CS and BRRS represent different points along the spectrum of PHTS. MAN in a background of LT and/or CCH are distinctive findings in this syndrome, and should alert pathologists to notify clinicians of the possibility of PHTS.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.