
Abstract
Background:
Subclinical hyperthyroidism is usually associated with Graves' disease or toxic nodular goiter. Here we report a family with hereditary subclinical hyperthyroidism caused by a constitutively activating germline mutation of the thyrotropin receptor (TSHR) gene.
Methods:
The proband was a 64-year-old Japanese woman who presented with a thyroid nodule and was found to be euthyroid with a suppressed serum TSH. The nodule was not hot. Although antibodies to thyroid peroxidase and thyroglobulin antibodies were present, TSHR antibodies were not detected by TSH-binding inhibition or by bioassay. Two of her middle-aged sons, but not her daughter, also had subclinical hyperthyroidism without TSHR antibodies. Without therapy, the clinical condition of the affected individuals remained unchanged over 3 years without development of overt hyperthyroidism.
Results:
A novel heterozygous TSHR point mutation causing a glutamic acid to lysine substitution at codon 575 (E575K) in the second extracellular loop was detected in the three family members with subclinical hyperthyroidism, but was absent in her one daughter with normal thyroid function. In vitro functional studies of the E575K TSHR mutation demonstrated a weak, but significant, increase in constitutive activation of the cAMP pathway.
Conclusion:
Although hereditary nonautoimmune overt hyperthyroidism is very rare, TSHR activating mutations as a cause of subclinical hyperthyroidism may be more common and should be considered in the differential diagnosis, especially if familial.
Introduction

To verify hereditary nonautoimmune hyperthyroidism in patients presenting with these variable clinical features, genomic DNA sequencing analysis of the TSHR gene and subsequent in vitro functional assays are essential to demonstrate that any mutation found increases receptor constitutive activity. Because this condition is inherited in an autosomal dominant manner, molecular diagnostics are advocated in the possible family members. After diagnosis, despite clinical differences in expression, ablative therapy (surgery or radioiodine) is commonly required to achieve long-term remission.
In this report, we describe a Japanese family in which all affected adult members presented with asymptomatic subclinical hyperthyroidism. This condition was associated with a novel constitutively activated mutation (E575K) in the second extracellular loop of the TSHR.
Case Report
Patient and her family
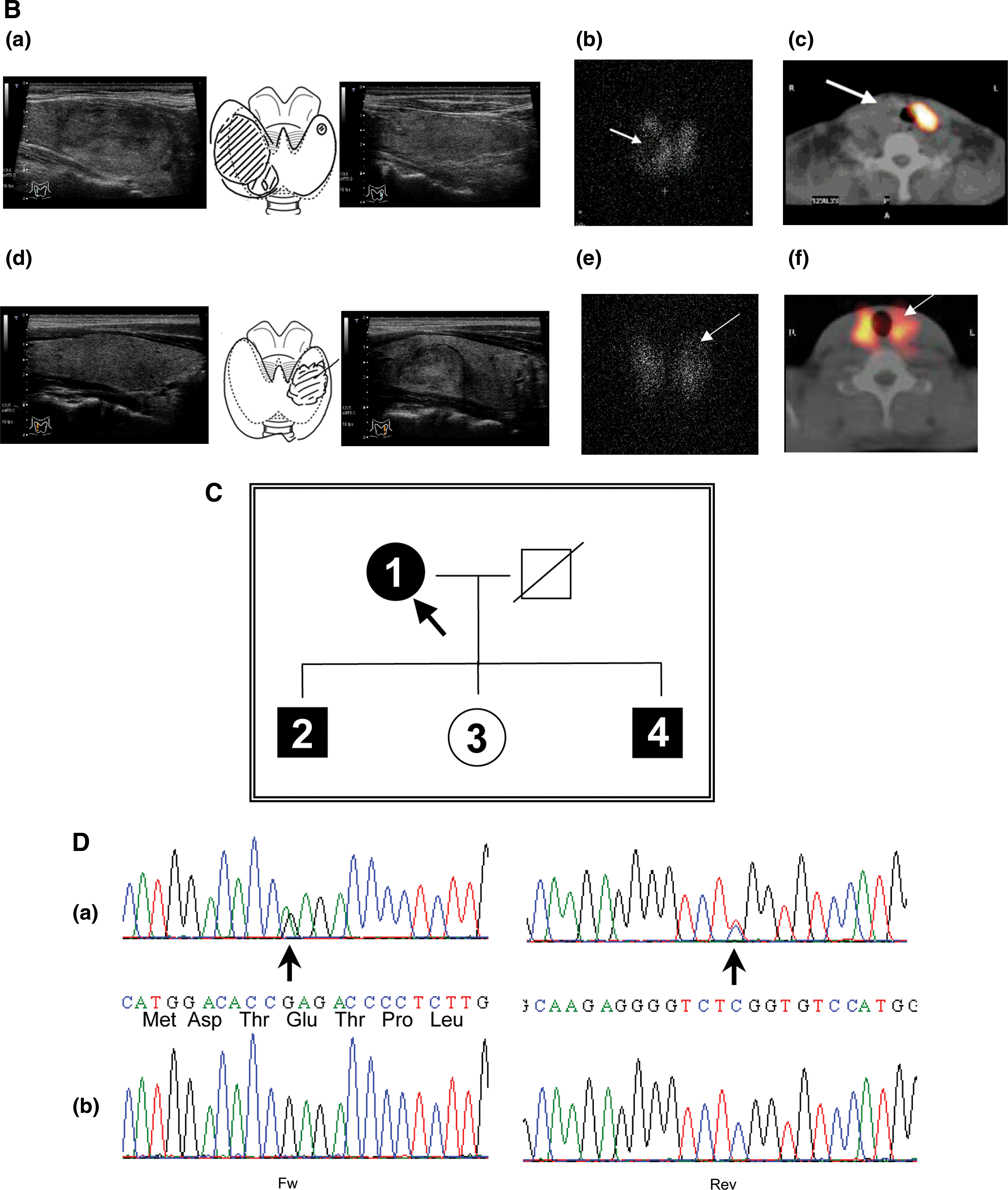
A 64-year-old Japanese woman consulted our hospital for the presence of a nodular lesion in the right lobe of the thyroid gland. Ultrasonography of the neck revealed a solid nodule with a maximum diameter of 4.2 cm and total thyroid volume of 40 mL (Fig. 1B-a and Table 1). She presented with subclinical hyperthyroidism (free thyroxine [FT4] 1.19 ng/dL, free triiodothyronine [FT3] 3.41 pg/mL, and TSH 0.032 mIU/L; see Materials and Methods for reference ranges). Anti-thyroid peroxidase (TPO) and anti-thyroglobulin (Tg) antibodies were positive, but anti-TSHR antibodies were negative in both a TSH-binding inhibition assay and a bioassay. Scintiscan imaging in the planar view showed radioiodine uptake in the normal thyroid tissue, but relatively faint uptake in the nodular lesion (Fig. 1B-b). This was further evaluated with single-photon emission computed tomography/computed tomography for the interpretation of inconclusive foci in planar view. The radioiodine uptake clearly localized to the normal thyroid tissue, and the nodule was therefore cold (Fig. 1B-c). Her two sons, but not her daughter, had normal levels of FT3 and FT4, and suppressed TSH levels with negative anti-TSHR antibodies (Table 1). Ultrasonography of the neck in her sons (members 2 and 4 in Table 1) revealed mild goiters, and that of member 4 revealed, additionally, a nodule with a maximum diameter of 2.4 cm (Fig. 1B-d). Scintiscan imaging in member 4 indicated diffuse radioiodine uptake and no abnormal uptake in the nodule (Fig. 1B-e, f). Specimens from the thyroid nodules in members 1 and 4 using fine needle aspiration indicated adenomatous nodules. The radioiodine uptake at 3 hours in the proband (member 1) and her two sons (members 2 and 4) were within the normal range (Table 1). No family members had symptoms of hyperthyroidism. Re-evaluation 3 years later of the proband confirmed the diagnosis of subclinical hyperthyroidism without progression to overt hyperthyroidism (FT4 1.25 ng/dL, FT3 3.00 pg/mL, and TSH 0.078 mIU/L). After 1.5 years of follow-up, subclinical hyperthyroidism persisted in member 4 (FT4 1.07 ng/dL, FT3 3.20 pg/mL, and TSH 0.089 mIU/L). There was no tendency toward progressive enlargement of the goiter and nodular lesions.
Normal levels: FT4, 0.70–1.60 ng/dL; FT3, 1.70–3.70 pg/mL; TSH, 0.30–5.00 μIU/mL; TBII, <1.9 IU/L; TSAb, <180 %; TgAb, <39.9 U/mL; TPOAb, <27.9 U/mL; Tg, <35 ng/mL.
RAIU in 3 hours, normal range 5.6%–15.8 %.
Normal thyroid volume, 5–20 mL.
F, female; FT3, free triiodothyronine; FT4, free thyroxine; M, male; NT, not tested; RAIU, radioiodine uptake; TBII, TSH-binding inhibitory immunoglobulins; Tg, thyroglobulin; TPO, thyroid peroxidase; TSAb, thyroid-stimulating antibody.
Materials and Methods
Thyroid function analysis and thyroid volume
Concentrations of serum TSH, FT3, and FT4 were measured with a chemiluminescent immunoassay (Architect i2000; Abbot Japan, Tokyo, Japan). The reference ranges used for serum TSH, FT3, and FT4 were 0.30–5.00 mIU/L, 1.70–3.70 pg/mL, and 0.70–1.60 ng/dL, respectively. Thyroid-stimulating antibodies were measured by a commercial bioassay kit (Yamasa Co., Chiba, Japan). Serum TSH-binding inhibitory immunoglobulins, anti-Tg antibodies, anti-TPO antibodies, and Tg were measured with an electro-chemiluminescent immunoassay (ECLusys TRAb, ECLusys Anti-Tg, ECLusys Anti-TPO, and ECLusys Tg; Roche Diagnostic, Mannheim, Germany). Total thyroid volume and the size and pattern of thyroid nodule were measured using ultrasound diagnostic equipment, as reported previously (17).
DNA sequencing
Genomic DNA was extracted from peripheral leukocytes with GenTLE (Takara, Kyoto, Japan). Exon 10 of the TSHR encoding the entire intracellular and transmembrane regions and part of the proximal extracellular domain of the TSHR was amplified by the polymerase chain reaction (PCR) using High Fidelity PCR Master (Roche Diagnostics) as described previously (23). Direct sequencing of PCR products was performed using the Bigdye terminator v1.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) and an automatic ABI 3130 sequencer (Applied Biosystems). The present study was approved by the ethics committee of Kuma hospital, and informed consent was obtained from the patient and her family members for the use of samples for research purposes.
Site-directed mutagenesis and functional expression of human TSHR
The E575K mutation was introduced into the wild-type TSHR cDNA in the vector pcDNA5/FRT (Invitrogen, Carlsbad, CA) using the QuickChange site-directed mutagenesis kit (Stratagene, San Diego, CA). The mutation was confirmed by nucleotide sequencing. Plasmids were transfected into Flp-In-CHO cells (Invitrogen) using Fugene HD (Roche Diagnostics). Because of difficulty in obtaining stable transfectants with hygromycin selection, we studied transiently transfected cells Flp-In-CHO cells. All cells were cultured in Ham's F12 medium supplemented with 10% fetal calf serum, penicillin (100 U/mL), gentamycin (50 μg/mL), and fungizone (2.5 μg/mL). Cells were passaged the day after transfection and tested 1 day later.
Flow cytometry
Chinese hamster ovary (CHO) cells were harvested from six-well plates using 1 mM ethylenediaminetetraacetic acid and 1 mM ethyleneglycoltetraacetic acid (EGTA) in phosphate-buffered saline (PBS). After washing twice with PBS containing 10 mM HEPES (pH 7.4), 2% fetal bovine serum, and 0.05% NaN3, the cells were incubated for 30 minutes at room temperature in 100 μL of the same buffer containing 1 μg of either normal mouse IgG or murine mAb 2C11 (Morphosys, Raleigh, NC). After rinsing, the cells were incubated for 45 minutes with 100 μL fluorescein isothiocyanate-conjugated goat anti-mouse IgG (1:100) (Caltag, Burlingame, CA), washed, and analyzed using a Becton-Dickinson flow cytofluorimeter (Franklin Lakes, NJ). Cells stained with propidium iodide (1 μg/mL final concentration) were excluded from analysis.
Cultured cell cAMP assays
CHO cells transiently expressing the TSHR were transferred into 96-well plates. For bioassay, the culture medium described above was replaced with F12 medium supplemented with 1 mM isobutyl methylxanthine, 10 mM HEPES, and, where indicated in the text, bovine (b) TSH (Sigma-Aldrich, St. Louis, MO). Mock tranfections with empty vector were included as controls. After 60 minutes at 37°C, the medium was aspirated and intracellular cAMP was extracted with 0.2 mL 95% ethanol. The extracts were evaporated to dryness and resuspended in 0.1 mL of PBS (pH 7.5), and samples (12 μL) were assayed using the LANCE cAMP kit according to the protocol of the manufacturer (PerkinElmer, Shelton, CT). Specific constitutive activity (SCA) (24) was calculated as (cAMP in TSHR-transfected cells−cAMP in control vector-transfected cells)/(flow cytometric geometric mean fluorescence in TSHR-transfected cells−fluorescence in control vector-transfected cells). SCA values were adjusted in proportion to that of the wild-type TSHR, which was normalized to a value of 1. SCA values were statistically analyzed using the Mann–Whitney Rank Sum test for non-normal values (SigmaPlot9; Systat, San Jose, CA).
Results
Identification of TSHR gene mutation
Sequence analysis from peripheral leukocytes of the proband (member 1) showed a heterozygous single base substitution at codon 575 of TSHR (GAG→AAG; E575K), located in the second extracellular loop (Fig. 1A and D). Sequence analysis of genomic DNA in members 2 and 4 showed an identical heterozygous guanine to adenine transition at position 1729; however, it was absent in member 3.
Functional studies of the novel mutant receptor
Functional studies of TSHR-E575K and the wild-type TSHR were performed in transiently transfected CHO cells. TSHR-E575K responded vigorously to TSH stimulation, with an intracellular cAMP increase similar to that of the wild-type TSHR (EC50s of ∼0.2 mU/mL) (Fig. 2). However, constitutive activity in the absence of TSH was increased with TSHR-E575K relative to the wild-type TSHR.

TSH stimulation of CHO cells transiently transfected with plasmids expressing the wild-type TSHR or TSHR-E575K. Mock transfections were performed with empty vector. Cells were stimulated for 1.5 hours with the indicated concentrations of TSH. Data represent the mean ± range of intracellular cAMP levels in duplicate wells of cells. The data shown are representative of three experiments with similar results.
To assess more accurately the ligand-independent activity of TSHR-E575K, we determined its SCA by adjusting cAMP values relative to the level of receptor expression on the cell surface as determined by flow cytometry. TSHR-E575K expressed well on the cell surface, though to a lesser degree than the wild-type TSHR (Fig. 3A). Normalizing the wild-type TSHR SCA to a value of 1 to facilitate inter-assay comparison; in three separate experiments the SCA of TSHR-E575K was significantly greater than the wild-type TSHR (2.1-, 1.8-, and 3.3-fold; mean 2.42 ± 0.46 standard error of the mean; p < 0.002) (Fig. 3B).
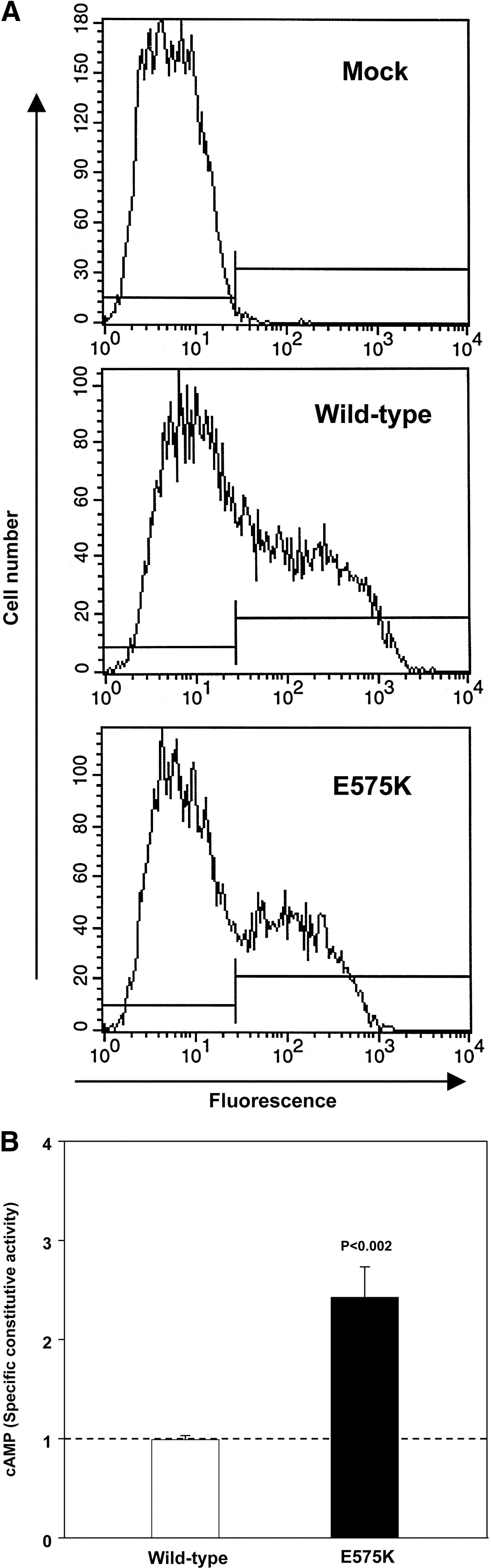
Increased specific constitutive activity of the E575K mutation.
Discussion
We report a family with hereditary nonautoimmune subclinical hyperthyroidism caused by a novel TSHR germline mutation (E575K). Compared to previous reports of this rare condition, the unique feature of our family is the mildness of the hyperthyroidism and its late presentation in all affected members. Indeed, the proband presented at age 64, and the disease was diagnosed only because of the concomitant presence of a very common condition, a thyroid nodule. The coldness of the nodule together with a suppressed serum TSH level excluded the preliminary suspicion of toxic nodular goiter. The difficulty in diagnosis was also compounded by the presence of autoimmune thyroiditis, as evident by serum autoantibodies to TPO and Tg. However, the absence of TSHR autoantibodies, determined by both bioassay and TSH-binding inhibition assay, made a diagnosis of Graves' disease highly unlikely. The activating TSHR mutation E575K, confirmed by in vitro functional analysis and segregating with the affected family members, provides a clear explanation for the diagnostic conundrum.
In vitro, the TSHR with the E575K mutation had a small but statistically significant increase in SCA with a cAMP value 2.4-fold greater than that of the wild-type TSHR. TSHR-E575K responded normally to TSH stimulation although its level of expression on the cell surface was slightly less than the wild-type TSHR (Figs. 2 and 3A), consistent with previous reports of constitutively activated TSHR mutations (25). To our knowledge, the E575K mutation is the second constitutively activating mutation in the second extracellular loop of the TSHR gene. Somatic (26) and germline (9,27) mutations at another codon in this loop (I568T and I568V) have been identified. Of interest, while the residue at codon 568 is highly conserved in the corresponding regions of other G protein-coupled receptors, E575 is not conserved. TSHR mutations leading to constitutive activation are often highly conserved in the G protein-coupled receptor family (28,29). In addition, the TSHR is more mutable than many other members of the G protein-coupled receptor family (28,30), generating more pathogenic mutations at various residues, including the novel mutation detected in individuals in the present report.
Nonautoimmune hyperthyroidism caused by an inherited mutation of the TSHR gene manifests at a variable age. A delayed and progressive onset of hyperthyroidism with mild clinical manifestations has previously been observed (1,2,11,14,17). However, to our knowledge, there are no prior reports on familial nonautoimmune hyperthyroidism with all affected individuals presenting in adulthood with subclinical hyperthyroidism. Even at the age of 67 the proband remains clinically asymptomatic and her thyroid function is within the normal range other than a suppressed TSH level. Whether the hyperthyroidism in her two sons with TSHR-E575K continues to be subclinical remains to be seen, although the mild and indolent progression of disease in their mother suggests that they may not require therapy in the future.
Analysis of genotype–phenotype correlations in nonautoimmune hyperthyroidism have shown no consistent relationship between in vitro activity of the mutant TSHR and the clinical course of the disease (31), suggesting that other genetic, epigenetic, and environmental modifiers, including iodine intake, might also affect clinical features. Although the prevalence of autonomous thyroid nodules causing hyperthyroidism is very low in Japan, where iodine intake is sufficient, the frequency of constitutively active somatic TSHR mutations in patients with this condition is similar to that in iodine-insufficient areas (32,33). In another family with hereditary nonautoimmune hyperthyroidism in Japan, the hyperthyroidism in the affected members was well controlled with inorganic iodine (17). Therefore, although the subclinical hyperthyroidism and the delayed presentation of the family in the present report is consistent with the small increase in TSHR-E575K constitutive activity, the disease may have been ameliorated by their living in a region with sufficient iodine intake.
The clinical importance of the family in the present report is to emphasize the difficulty in diagnosing hereditary nonautoimmune hyperthyroidism, a rare disease, when affected family members have subclinical hyperthyroidism, manifest only by a suppressed TSH. A suppressed TSH without elevated thyroid hormone levels occurs in evolving toxic nodular goiter and at an early stage of Graves' disease, both very common disorders. The proband in the present study had a large thyroid nodule as well as autoimmune thyroiditis. The correct diagnosis was only suspected upon exclusion of the latter two diagnoses by the precise location of radioiodine uptake by single-photon emission computed tomography/computed tomography analysis and by the absence of TSHR autoantibodies. Careful interview of all family members and examination of their thyroid function are practical before sequence analysis of the TSHR gene. There has been no progression to overt hyperthyroidism in members of our family within 3 years of follow-up. If clinical hyperthyroidism does develop, ablative therapy will be required. The parents of the proband died without a history of overt thyroid disease, but further genetic examination of the kindred in this family may reveal other affected members who will require careful monitoring.
In summary, we have identified a Japanese family with hereditary nonautoimmune hyperthyroidism. Unlike in previous families with this condition, the disease in all affected members has remained subclinical even into adulthood and advanced age. A novel TSHR mutation (E575K) is responsible for a small but significant increase in constitutive activity. A diagnosis of familial subclinical hyperthyroidism may be obscured by other thyroid diseases occurring more frequently in the general population. We suggest that this condition may be more common than presently believed and that families with overt hereditary hyperthyroidism may represent the tip of the iceberg.
Footnotes
Acknowledgment
This work was supported by NIH Grant DK19289 (B.R.).
Disclosure statement
The authors declare that no competing financial interests exist.