
Abstract
Background:
Germ-line mutations of RET proto-oncogene are the known cause of hereditary medullary thyroid carcinoma (MTC), which account for approximately 25% of all MTC cases and occur as multiple endocrine neoplasia type 2 syndromes. Here, we present the first comprehensive genetic screening and analysis of MTC among Iranian families.
Methods:
A total of 55 patients with MTC (male to female ratio = 1:1.6; average age of disease onset = 33 ± 13 years) from 53 independent families participated in this study. All of the patients had undergone total thyroidectomy between 1999 and 2006, and 51 of them were clinically characterized as apparently sporadic cases. Genomic DNA samples were obtained and following highly-specific polymerase chain reaction amplification of the 6 RET key exons (10, 11, 13, 14, 15, and 16) were subjected to direct DNA sequencing without a requirement for a purification step.
Results:
Sequence analysis revealed that 9 (17.6%) of the apparently sporadic cases (from 8 kindreds) carried an RET germ-line mutation. Of the seven different mutations identified among all of the families studied, five were in the cysteine codons, with Cys634Arg having the highest prevalence (45.5%) among the afflicted families. Mutation carriers have an earlier age of onset (21 ± 6) versus the sporadic cases (37 ± 12).
Conclusions:
This is the first comprehensive genetic screening and analysis of MTC among Iranian families. The results further confirm the need and advantages of DNA sequencing for identification of hereditary MTC cases. There does not seem to be a meaningful correlation between single nucleotide polymorphism patterns and the average age of disease onset. Geographical distribution of the sporadic cases, however, shows a significant concentration toward the Northern regions of the country, noticeably the provinces situated directly to the south of the Caspian Sea.
Introduction
Germ-line missense mutations of RET proto-oncogene are the known cause of all hereditary MEN2 variants (3,4). RET gene (RET), which is located on chromosome 10 near the centromere (10q11.2), encodes a transmemebrane receptor tyrosine kinase protein that is primarily expressed in neural crest and urogenital precursor cells. RET molecule contains an extracelluar, a transmembrane, and two intracellular tyrosine kinase domains. Traditionally, elevated serum calcitonin levels, both basal and following pentagastrin stimulation, were used as an indicator for C-cell hyperfunction and MEN2 carrier status. However, molecular genetic studies have provided a reliable method for identification of RET mutations and established a strong correlation between the genotypes and MEN2 phenotypes (2,5–7). Mutations in exons 10 and 11 (e.g., Cys620Tyr and Cys634Arg), affecting the cysteine-residue encoding codons within RET extra-cellular domain, play a major role in causing MEN2A. These mutations often lead to a gain-of-function in RET tyrosine kinase signaling as a result of auto-dimerization of two RET molecules. Some other mutations in exons 10 and 11 as well as mutations in exons 13, 14, and 15 (which encode the RET intra-cellular domain) could lead to FMTC. By contrast, MEN2B is almost exclusively related to Met918Thr mutation in exon 16 that also leads to a gain-of-function as a result of substrate alteration (8 –13). Therefore, analysis of the 6 RET key exons (no. 10, 11, 13, 14, 15, and 16) is a very reliable method in order to differentiate hereditary MTC cases from sporadic ones (14).
Before the present work, the available published data on the rate, distribution, and mutation analysis of MTC among the Iranian population were very limited. In a retrospective study, Larijani and colleagues analyzed the medical records of five major tertiary referral centers in the Iranian capital, Tehran. The statistical analysis of 1,177 thyroid carcinoma cases, from a 14-year period (1980–1994), showed that only 42 patients (3.6%) were diagnosed as MTC, with a female to male ratio of 1:1.1 (15). The molecular study of patients with MTC in Iran has been limited to a report by Hedayati et al., in which examination of RET exons 10 and 11 in 57 MTC cases (with a female to male ratio of 1.2:1) by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) led to identification of only four germ-line mutation carriers (16).
Here, we present the first comprehensive molecular analysis of patients with MTC among the Iranian population. After a thorough genetic counseling phase, a total of 55 patients with MTC from 53 independent kindreds were examined by direct DNA sequencing of all 6 RET key exons:10, 11, 13, 14, 15, and 16. This led to a reliable strategy for identification of MTC germ-line mutation carriers, exact genotyping of all index patients, and a prelude to molecular screening of their first-degree relatives. Molecular characterization of the asymptomatic germ-line mutation carriers provided a highly significant preventive opportunity by their timely referral for prophylactic thyroidectomy.
Materials and Methods
Patients
Fifty-five patients with MTC from 53 independent families were referred from three main specialized medical centers in the capital city, Tehran (TUMS Endocrinology and Metabolism Research Center, EMRC, at Shariati Hospital; Shahid Beheshti University's Endocrine Research Center at Taleghani Hospital; and Endocrinology and Metabolism Research Center at Vali-Asr Hospital), which deal with most MTC cases in Iran. These cases were diagnosed and treated during an eight-year period from 1999 to 2006.
MTC or C-cell hyperplasia was initially detected by elevated serum-calcitonin levels, followed by fine needle aspiration. After surgical removal of the thyroid gland, the diagnosis of MTC was further confirmed by histopathology. The initial MEN2 variant type assignments were made based on clinical manifestations, medical records, including biochemical data and histopathological features, and family pedigrees.
Genetic counseling
All of the index patients were subjected to genetic counseling, during which background of the disease, the aim of the study, and the potential risk for other family members were discussed. An informed consent form was obtained from each patient and approved by TUMS ethics committee in accordance with institutional guidelines and national regulations.
After molecular analysis, a formal report containing the results was prepared for each family/patient, by addressing respective endocrinologists. After identification of the families with RET germ-line mutation carrier/s, a second genetic counseling session was held, during which arrangements were made for collecting blood samples from the first-degree relatives of the index patients. In case of detecting an asymptomatic germ-line carrier during the screening process, he/she was referred for prophylactic thyroidectomy.
DNA extraction
Using a modified salting-out technique, genomic DNA was extracted from peripheral blood lymphocytes of ethylenediaminetetraacetic acid-anticogulated samples. Briefly, 500 μl of each blood sample was mixed with 1 mL of RBC-lysis buffer (0.32 M sucrose, 10 mM Tris-HCl, pH 8.2, 5 mM MgCl2, and 1% v/v Triton X-100), incubated on ice for 15 min, and centrifuged at 10,000 rpm for 2 min. After discarding the supernatant, the same procedure was repeated twice before addition of 300 μl of WBC-lysis buffer (0.45 M NaCl, 10 mM Tris-HCl, pH 8.2, and 2 mM ethylenediaminetetraacetic acid), 20 μl of 10% sodium dodecyl sulfate, and 10 μl of proteinase K (20 mg/mL), followed by incubation at 55°C for 2 h. gDNA was precipitated by addition of 100 μl of 5M NaCl and isopropanol (1v/v), fished out using a thin glass rod, washed with 70% EtOH, and dissolved in 1 × TE buffer. The quality of gDNA samples and their quantification were determined by agarose gel electrophoresis and UV spectrophotometry (A260/A280).
PCR amplification of RET key exons
Forward (F) and reverse (R) primers for RET exons 10, 11, 13, 14, 15, and 16 were designed based on analysis of DNA sequences as well as some previous studies (17–20): exon 10, RET10-F2 (5′-CTATGCTTGCGACACCAGTTG-3′) and RET10-R2 (5′-CTCCTGGGTGGAGTAACAGAG-3′); exon 11, RET11-F2 (5′-CAGAGCATACGCAGCCTGTAC-3′) and RET11-R1 (5′-GCCTCGTCTGCCCAGCGTTG-3′); exon 13, RET13-F1 (5′-AGAAGCCTCAAGCAGCATCGTC-3′) and RET13-R1 (5′-AGGAGCAGTAGGGAAAGGGAGA-3′); exon 14, RET14-F1 (5′-GAAGACCCAAGCTGCCTGAC-3′) and RET14-R1 (5′-GGCTAGAGTGTGGCATGGTG-3′); exon 15, RET15-F1 (5′-GGTCTCACCAGGCCGCTAC-3′) and RET15-R2 (5′-TCGGTATCTTTCCTAGGCTTC-3′); exon 16, RET16-F1 (5′-AGGGATAGGGCCTGGGCTTC-3′) and RET16-R1 (5′-CCAGCCATTTGCCTCACGAAC-3′).
PCRs were performed in 25-μL mixtures containing 50–100 ng of template gDNA, 0.2–0.4 μM of each primer, 1 × PCR buffer with 1–2 mM of Mg2+, and 0.625 U of Taq polymerase (Roche Diagnostics). The concentrations of Mg2+ (CinnaGen Inc.), dNTPs (CinnaGen Inc.), and template gDNA was specifically worked out for each exon. PCR amplification was performed with a pre-heating cycle of 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 60°C–62°C for 30 s, extension at 72°C for 40 s, and a final extension cycle at 72°C for 7 min. Afterward, 5 μl of each PCR was analyzed by gel electrophoresis on 1% agarose, and a portion of each reaction was used for direct DNA sequencing.
DNA sequence analysis
DNA sequencing reactions were performed by BigDye Terminator Cycle Sequencing kit (Applied Biosystems) in a 20 μl final volume, using 10 ng of each PCR product as template and 3.2 pmol of the respective primer per reaction. The reactions were run on an automated ABI Sequence Analyzer 3130xl (Applied Biosystems). The PCRs for each sample were initially sequenced using the forward primers. In the case of germ-line mutation carriers, however, the results were also re-confirmed by another round of PCR and DNA sequencing using the respective reverse primers for further assurance. The ABI chromatograms were each carefully examined using Sequence Scanner (Applied Biosystems) before subjecting them to alignment analysis with Lasergene MegAlign software (DNASTAR).
Restriction fragment length polymorphism
In order to detect the Cys634Arg mutation (TGC → CGC) by a restriction endonuclease, the PCR products of RET exon 11 (454 bp) were first purified and quantified, as mentioned earlier, and were then digested by Hha I (CinnaGen Inc.) following the manufacturer's instructions. Digested fragments along with nondigested ones (as negative controls) were analyzed on 2% agarose gels.
Results
MTC patients characterization and demographics
A total of 55 patients with MTC, with a female to male ratio of 1.6:1, from 53 kindreds (i.e., unrelated families) were identified by reviewing the medical records from major referral centers in the Iranian capital city, Tehran, and enrolled for this study (Table 1). The average age of disease onset was 33 (±13) years. All of these cases had undergone total thyroidectomy and were still alive during the course of study. Based on reviewing the clinical records and information obtained during the genetic counseling sessions, only four patients from four families were characterized with MEN2 phenotypes: no. 2 and 31/2, MEN2A; no. 26, FMTC; and no. 53, MEN2B. The remaining 51 patients, representing 49 independent families, were considered as apparently sporadic cases.
Index patients' phenotypes: aMEN2A; bFMTC; cMEN2B.
NC, not confirmed; MTC, medullary thyroid carcinoma; MEN2A, multiple endocrine neoplasia type 2.
Molecular analysis of the RET proto-oncogene key exons
PCR set-ups
One of the goals of the present work was to devise an efficient, time-saving, and cost-effective strategy for amplification and sequencing of the 6 RET key exons (10, 11, 13, 14, 15, and 16). This was achieved by working out a detailed PCR-setup for highly specific amplification of each exon, as well as efficient primer usage, without the need for either a gel- or PCR-purification step before DNA sequencing (Table 2). An example of this approach, how the specific conditions were worked out for RET exon 11, is shown in Figure 1 (also see Materials and Methods for details on gDNAs quality and sequencing reactions). It should be noted that except for Lane 1, which was the first amplification reaction from the 6-exon PCR set, each lane is a representative of a set of reactions focusing on a specific PCR condition, such as the amount of template gDNA, annealing temperature, and Mg2+ or dNTPs concentrations (Fig. 1).
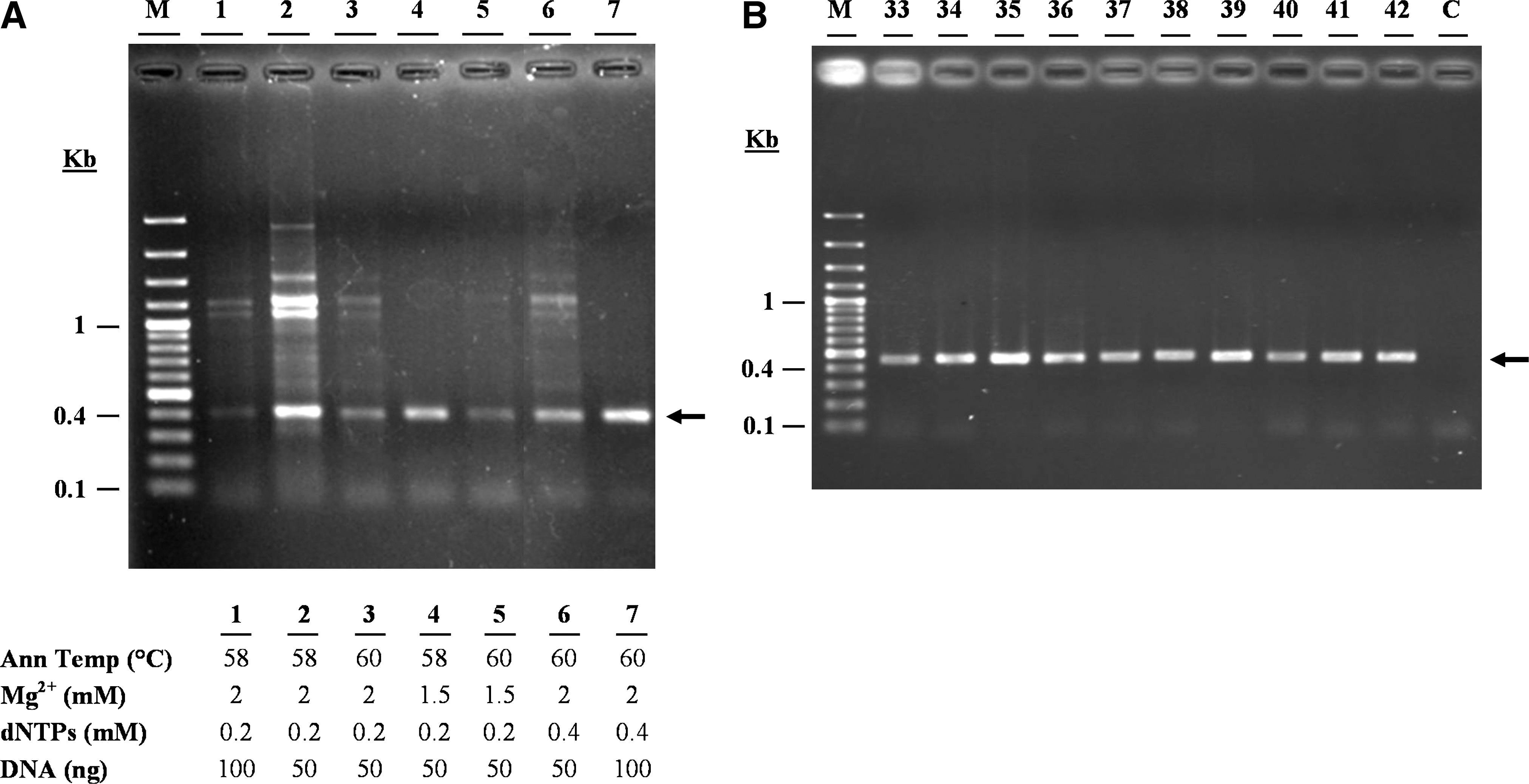
RET exon 11 polymerase chain reaction (PCR) set-up.
Reaction volume: 50 μl.
Concentration of each of dNTP.
F, forward primer; R, reverse primer; PCR, polymerase chain reaction.
Characterization and distributions of RET mutations
Among the 53 kindreds studied here, 11 (20.7%) were identified as carrying RET germ-line mutation carriers (Table 3). The 4 families (no. 2, 26, 31, and 53) with earlier MEN2 phenotype assignments matched their predictive genotypes. There were 13 patients who were index mutation-carriers with an average age of disease onset of 20.5 years old (±6), ranging from 10 to 31 years. Each of these identified mutation carriers harbored only a single germ-line missense point-mutation. Although one cannot rule out the possibility of changes within other RET exons, we did not find any evidence of double mutations or deletion/insertions within the 6 key exons examined here. Overall, a total of 7 different heterozygote missense point-mutations were identified in RET exons 10, 11, 14, and 16 (Fig. 2). There were no mutations in exons 13 and 15. The most affected part of the RET molecule came from exons 11 (seven families, 63.6%) and 10 (two families, 18.2%), which encode the cysteine-rich regions of the extra-cellular domain, followed by exons 14 and 16 (one family each, 9.1%), which encode the tyrosine kinase regions of the intra-cellular domain. In fact, 9 of the 11 afflicted families carry germ-line mutations in cysteine codons.
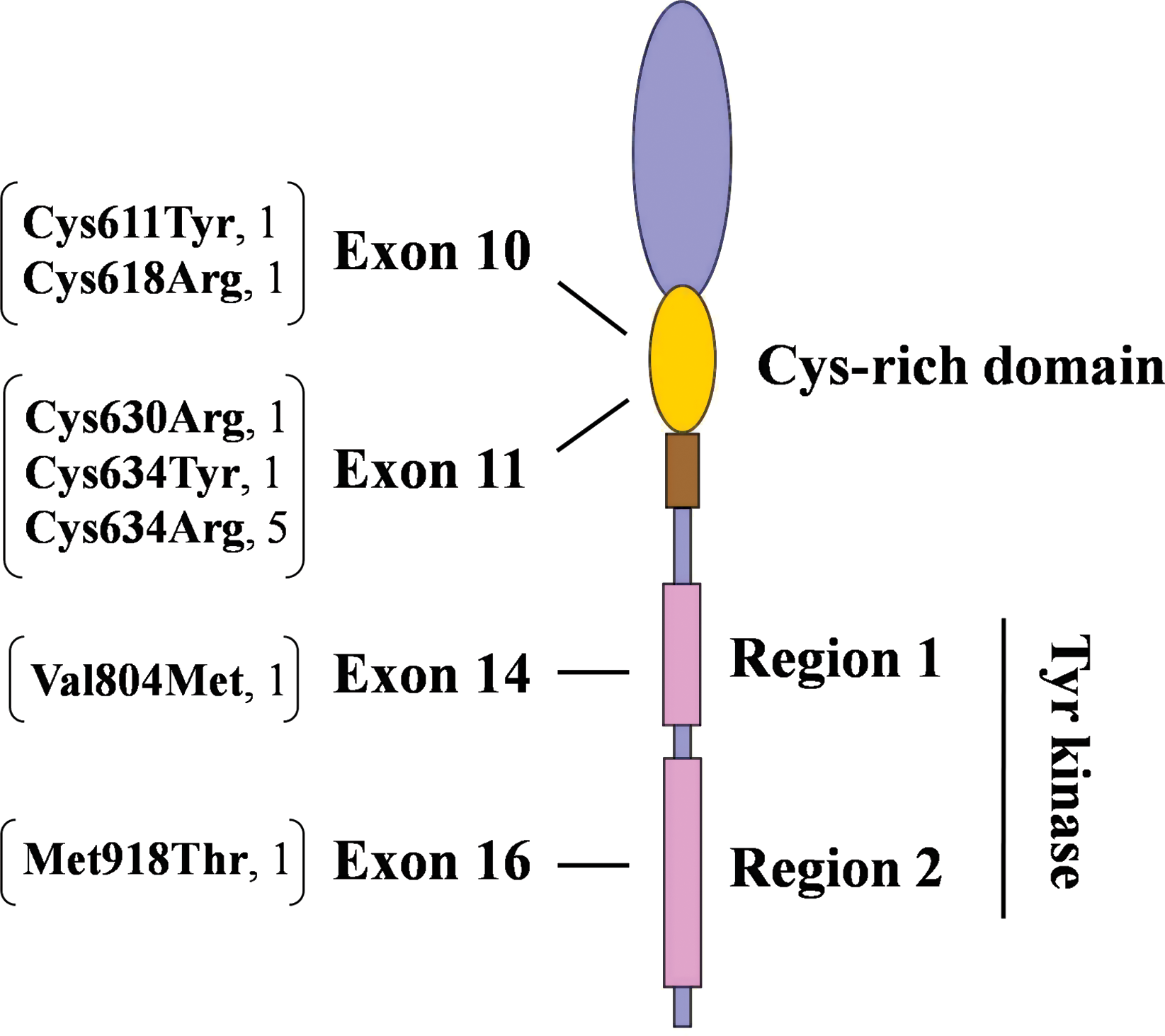
Distribution of RET germ-line mutations. The brackets show the affected codons and resulting amino acid changes, followed by the number of kindreds identified. Color images available online at
Notice that phenotypes assignments were done before molecular characterization. Question marks for patients 31/1 and 31/3 indicate suspicion for being a mutation carrier based on their family history.
FMTC, familial medullary thyroid carcinoma.
The most prevalent mutation, Cys634Arg (TGC → CGC), which is indicative of MEN2A, was seen in 7 carriers (53.9%) from 5 (45.5%) kindreds. In fact, apart from cases with assigned MEN2 phenotypes, the Cys634Arg mutation was found in 4 out of 49 (i.e., 8.2%) apparently sporadic MTC cases. The remaining six mutations (Cys611Tyr, Cys618Arg, Cys630Arg, Cys634Tyr, Val804Met, and Met918Thr) each represented a single index patient from one (9.1%) mutation-carrier kindred. Surprisingly, the mutation carrier with the youngest age of disease onset (patient no. 5, 10 years) was genotyped as Val804Met (GTG → ATG), which is typically classified as level 1 (i.e., the lowest) risk level.
Single nucleotide polymorphism mapping and geographical distribution of MTC cases
In addition to characterization of germ-line mutations, DNA sequencing provided a complete and detailed genotype for each index MTC patient. Table 4 shows the distribution of known single nucleotide polymorphism (SNPs) among all 53 patients with MTC. Only one of the observed SNPs, codon 691 from exon 11, results in an amino-acid change: GGT → AGT; Gly691Ser. In addition to the SNPs listed on the table, we detected a potentially new SNP (AAG → AAA; Lys887Lys) at codon 887 from exon 15 in four members of kindred number 31, including two index patients: 31/1 and 31/2 (Fig. 3).
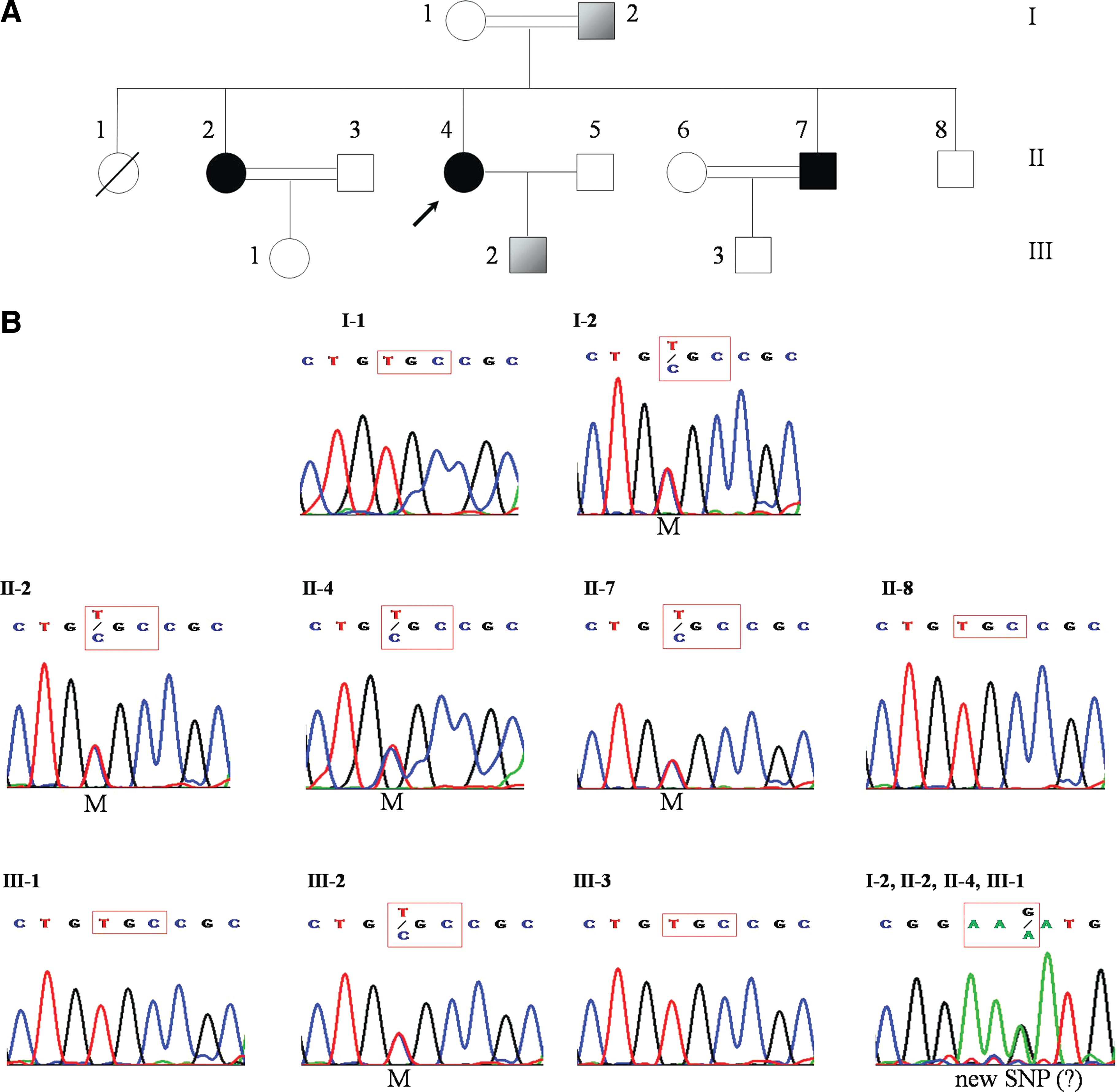
Pedigree analysis and sequencing results for kindred no. 31.
Bold numbers (2, 5–7, 11, 21, 26 31/1–3, 39, 51, and 53) indicate the RET germ-line mutation carriers.
Also contained AAG → AAA (K887K).
SNP, single nucleotide polymorphism; —, No SNP detected; P, heterozygote SNP; PP, homozygote SNP.
We found no apparent significant correlation between observed SNPs and patient gender or age of disease onset. By contrast, verified geographical distribution of the patients with MTC shows a significant concentration of sporadic cases toward the North and Northwestern regions of the country (Fig. 4).
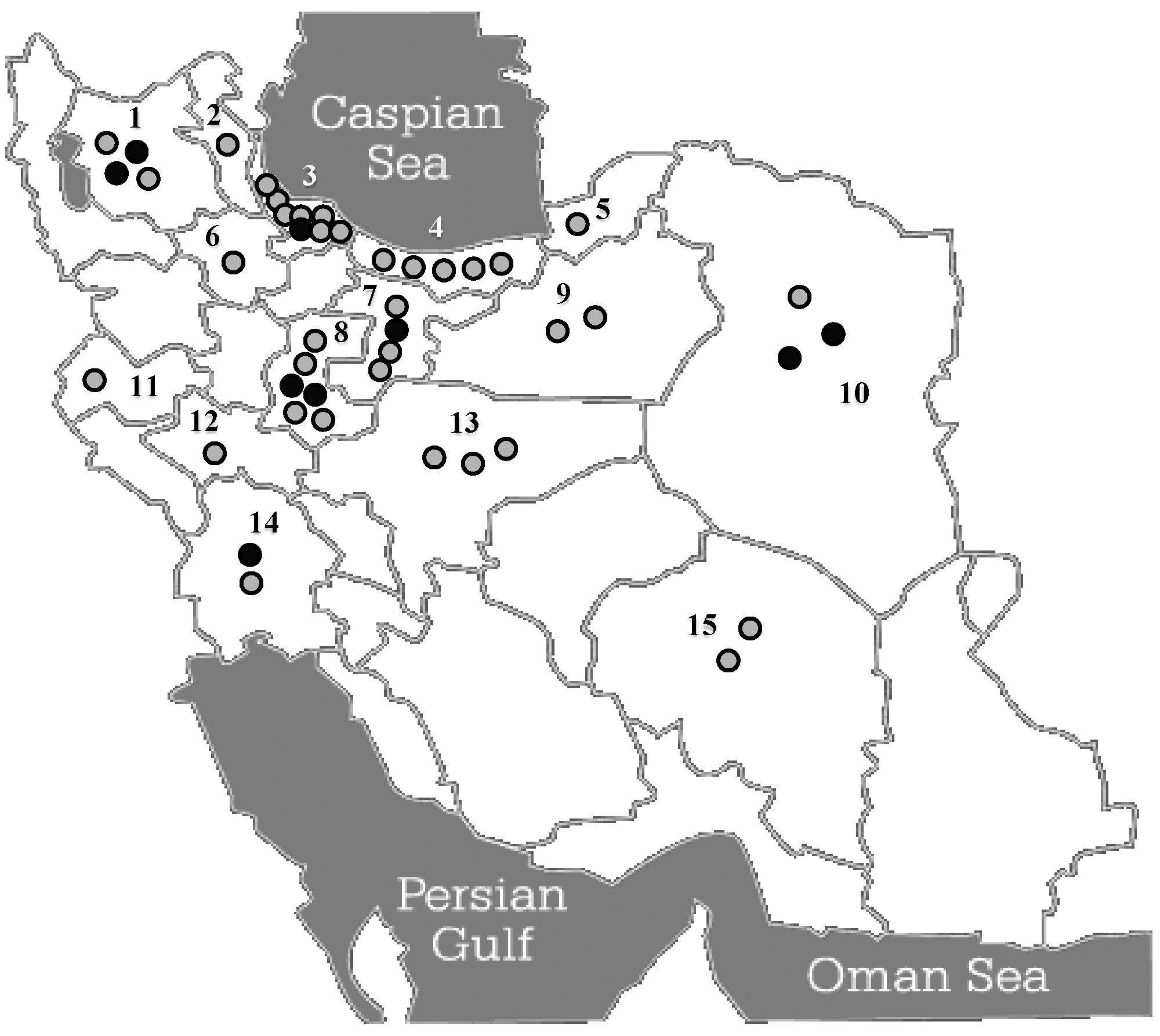
Geographical distribution of patients with MTC in Iran. The locations of the families harboring RET germ-line mutation carriers are shown with black circles, whereas those with sporadic cases are indicated by gray circles. The high concentration of sporadic cases in the two Northern provinces of Gilan and Mazandaran (labeled on the map by numbers 3 and 4, respectively) might be because of localized environmental factors. Numbers indicate various provinces as confirmed origins of patients with MTC: 1, East Azarbayejan; 2, Ardebil; 3, Gilan; 4, Mazandaran; 5, Golestan; 6, Zanjan; 7, Tehran; 8, Markazi; 9, Semnan; 10, Khorasan; 11, Kermanshah; 12, Lorestan; 13, Esfahan; 14, Khuzestan; and 15, Kerman.
Discussion
Considering the prevalence of MTC in Iran, based on the available current information and two previous studies spanning a 26-year period (1980–2005) (15,16), our sample size of 53 independent families, from a recent 8-year period (1999–2006), covers a very significant portion, if not the vast majority, of MTC cases in the country. To our knowledge, this is the first comprehensive genetic analysis and screening for MTC among the Iranian population. Sequencing of the 6 RET key exons resulted in identification of 13 germ-line mutation carriers from 11 independent families. Mutation carriers had an earlier age of MTC onset compared with the sporadic cases: 21 ± 6 versus 37 ± 12, respectively. Noticeably, both of these numbers are indicative of earlier ages of onset for MTC in Iran compared with the global averages [around the age of 30 for hereditary vs. the 6th decade of life for sporadic cases (4,5)].
The initial screening of the patients with MTC, based on reviewing their medical records and information obtained during the genetic counseling sessions, resulted in characterization of only four patients, from four independent families, with MEN2 phenotypes: two cases of MEN2A and one case of MEN2B and FMTC each, from families number 2, 31, 53, and 26, respectively (Table 3). Later on, molecular analysis of RET germ-line mutations confirmed the initial phenotypic assignments. It should be noted that none of the patients had exhibited the symptoms of Hirschsprung's disease. Two patients showed the characteristics of MEN2A: patient no. 2 carrying the Cys634Tyr mutation had hyperparathyroidism, and no. 31/2 carrying Cys634Arg mutation exhibited pheochromocytoma.
We also suspected two patients (31/1, and 31/3) as potential cases of MEN2A based on the assignment of MEN2A phenotype to another member (31/2) of the same kindred (Fig. 3A). However, these suspected cases lacked the proper clinical manifestations for such a diagnosis. Most notably, unlike patient no. 31/2, they had shown no evidence of pheochromocytoma. Nonetheless, as shown in Figure 3B, genetic screening revealed that both suspected patients carried the Cys634Arg germ-line mutation, which is the genetic hallmark of MEN2A.
Apart from the six cases discussed earlier, the remaining 49 independent patients with MTC had neither the clinical manifestations nor a family history indicative or pointing at MEN2 subtypes. Therefore, they were characterized as apparently sporadic cases. However, molecular analysis of the 6 key RET exons by DNA sequencing revealed that seven of these patients harbored single germ-line mutations.
Among the 53 families studied here, RET germ-line mutations were detected in 11 families (20.7%). The index patients in six (54.5%) of these families harbored a mutation in codon 634, with Cys634Arg (TGC → CGC) being the most frequent one. The Cys634Arg is the most frequent, and exclusive, mutation associated with MEN2A. There is also generally a strong correlation between mutations of codon 634 and the occurrence of pheochromocytoma and/or hyperparathyroidism as well as an increasing probability of developing these phenotypes with age (1 –3). Therefore, we concluded and recommended that all of the Cys634Arg carriers go through regular clinical follow-ups.
Despite the exact criteria of International RET Mutation Consortium for classification of FMTC (i.e., families with more than 10 carriers and affected members over the age of 50) (14), we classified one family (no. 26) as FMTC. Based on the clinical data, only four members of this family had developed MTC, and they all lacked other MEN2-associated manifestations. However, this is in line with a less rigid definition of FMTC and the recent trend advocated by the American Thyroid Association (2). Moreover, random genetic screening of two of the four members revealed that they both carried the rare germ-line mutation TGC → CGC (Cys630Arg) in exon 11, which is in agreement with FMTC characterization (21,22).
We detected a Val804Met mutation in RET exon 14 of a 10-year-old patient (family no. 5). In some reports, mutations in codon 804 have been reported to be associated with late age of MTC onset and low tumor aggressiveness (4,5,23). By contrast, another study reported a Val804Met carrier showing the symptoms of the disease at the age of six, who died 6 years later due to widespread metastases (24). Finally, the presence of pheochromocytoma and/or hyperparathyroidism in Val804Met carriers have been reported in a number of studies (25 –27). Patient no. 5 did not exhibit any other endocrine-related phenotypes. However, the presence of MTC at the age of 10 supports the previously proposed wide range for the age of disease onset among Val804Met carriers (28).
In approximately 95% of cases, MEN2B is associated with a methionine-to-threonine substitution in the intra-cellular domain of RET as a result of mutation in codon 918 of exon 16 (Met918Thr) (10 –12,29). We found this germ-line mutation in patient no. 53 (age of MTC onset: 14) who had already been characterized as MEN2B based on her medical records. Interestingly, both parents, who were over the age of 35, appeared healthy. Although we did not succeed in arranging for genetic screening of the parents, considering their ages, it is likely that the index patient is a de novo germ-line mutation carrier. The presence of de novo germ-line Met918Thr mutation has been reported by other investigators as well and, in fact, accounts for more than 50% of the cases (2,30,31).
In a previous study from Iran by Hedayati and colleagues, the investigators looked at RET exons 10 and 11 of 57 patients with MTC (collected within a 15 years period) by PCR-RFLP analysis. The authors reported finding of 4 RET germ-line mutation carriers (16). In a collaborative effort with Dr. Hedayati's laboratory and careful cross-referencing of patients' information, 22 of our MTC samples were from the same patients as the previous study. Among these patients, using the PCR-RFLP technique, Hedayati et al. had found only one patient carrying a Cys620Arg mutation by digesting the gDNA with BstU I. Surprisingly, by using DNA sequencing, however, we did not detect any germ-line mutation in this patient. Further, RET sequencing led to identification of 2 germ-line Cys634Arg carriers that had escaped detection by PCR-RFLP in the previous study. We repeated the molecular analysis of these patients by DNA sequencing of fresh samples, and the same results ensued. Further, subjecting our gDNA samples to PCR-RFLP using the restriction enzyme Hha I also provided the expected outcomes (Fig. 5). Nonetheless, such discrepancies confirm the reliability of DNA sequencing and its preference over RFLP in detection of RET germ-line mutations as also stated by the International RET Mutation Consortium (14).
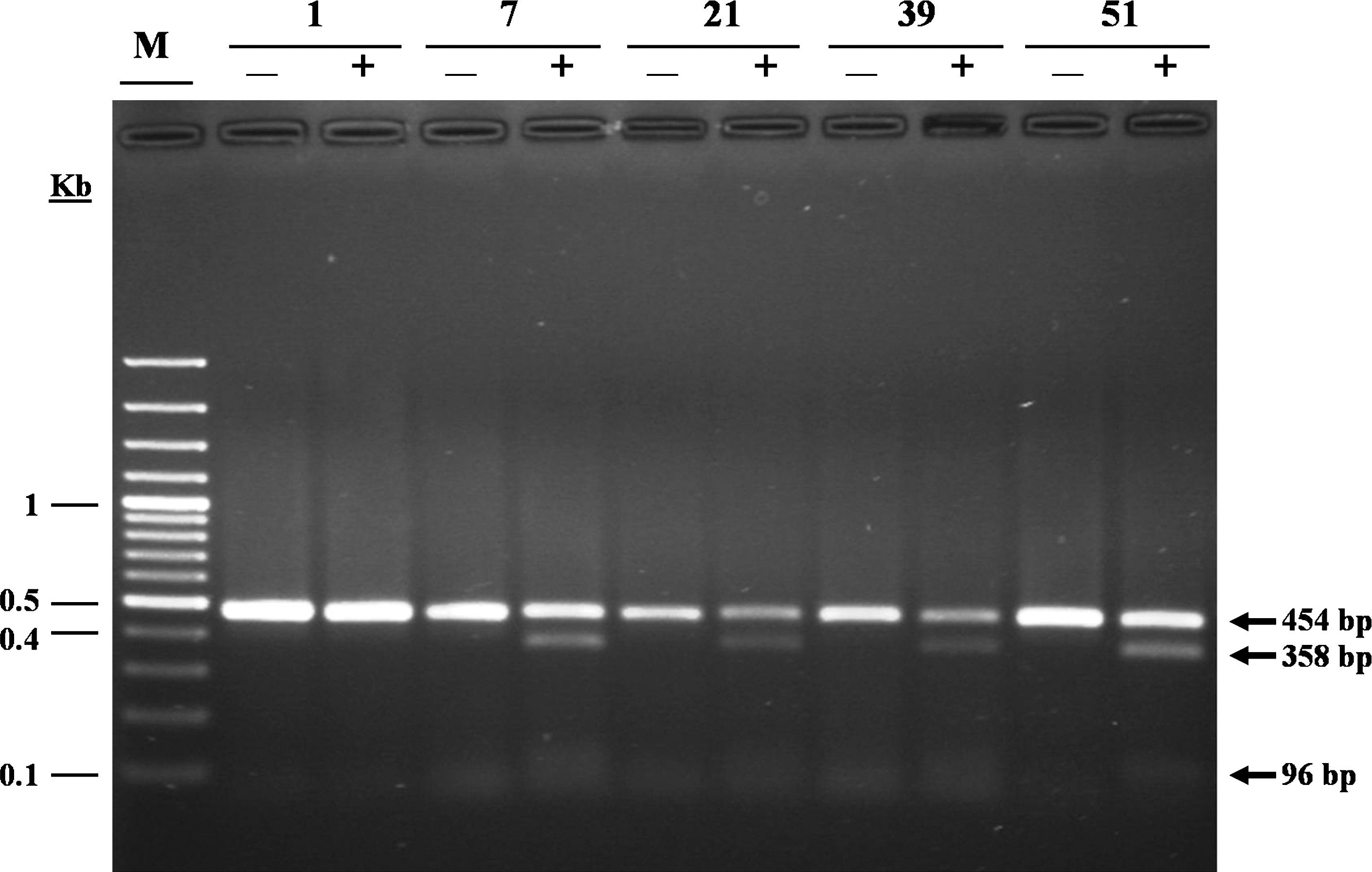
Restriction fragment length polymorphism (RFLP) analysis of Cys634Arg. Restriction digestion of 454-bp amplified product of RET exon 11, containing Cys634Arg, by Hha I results in formation of 2 DNA fragments (358 and 96 bp). Due to the lack of mutated allele, the PCR product from sample 1 does not get cut. By contrast, samples 7, 21, 39, and 51 from respective patients all contain the heterozygote mutation. Lanes: M, 100-bp DNA ladder; + and −, with and without the restriction enzyme, respectively.
In addition to identifying the germ-line mutations in an unbiased manner, another advantage of RET sequencing is that it provides a clear genotype, which includes an SNP map as well. We have obtained clear and complete genotypic profiles of the 6 RET key exons for our patients with MTC. Interestingly, we also detected a potentially new SNP, without amino acid substitution, in exon 15 (AAG → AAA; Lys887Lys) of four members of one family (Fig. 3). Whether this nucleotide change has any effect on MTC progression requires further investigation. There have been some contradictory results in different studies regarding the association of SNPs with a germ-line mutation and/or mean age of the disease onset (32 –40). Given our limited sample size, we did not find a statistically significant correlation between the SNP maps and patient gender and/or age of disease onset within our samples.
We found no specific geographical concentration of RET germ-line mutation carriers in Iran. By contrast, geographical distribution of sporadic cases shows a high concentration within the Northern regions, especially in the provinces of Gilan and Mazandaran, which are located by the Caspian Sea (Fig. 4). Although these regions are densely populated, considering the fact that they make up only about 8% of the entire population of the country (41), this might be suggestive of involvement of some environmental factors.
This study is the first comprehensive survey of patients with MTC in Iran. Despite the poor system of assorting data of patients with MTC, along with very low frequency of the disease, through concerted effort and teamwork, we managed to collect data from three major endocrinology and surgery departments in the country. We also set up a reliable step-by-step strategy for molecular analysis of RET proto-oncogene for patients with MTC in Iran. During the course of this study, by screening the first-degree relatives of identified hereditary cases among three of the 11 families containing index germ-line carriers, we were successful in detecting four asymptomatic germ-line mutation carriers and referred them for prophylactic thyroidectomy. Further follow-up of families carrying germ-line mutation is needed to improve their health state.
Footnotes
Acknowledgment
This work was supported in part by TUMS grant number 3320.
Author Disclosure Statement
No competing financial interests exist.