
Abstract
Background:
Oxidative stress associated with 3,3′,5-triiodo-
Materials and Methods:
The effect of T3 administration in the presence and absence of N-acetylcysteine (NAC) on cytosol-to-nuclear translocation of Nrf2 was evaluated, with inhibition of this process by NAC taken as evidence that the process was redox mediated. Male Sprague-Dawley rats weighing 180–200 g were given a single intraperitoneal dose of 0.1 mg T3/kg. Another group of rats were given the same dose of T3 and were also pretreated with NAC (0.5 g/kg) at 0.5 hour before T3 administration. Two other groups of rats received vehicle treatment and NAC, respectively. Following these treatments, rectal temperature of the animals, liver O2 consumption, serum and hepatic levels of 8-isoprostanes, and liver protein levels of Nrf2, Akt, p38, and thioredoxin (Western blot) were determined at different times up to 48 hours.
Results:
T3 administration induced a significant increase in the hepatic nuclear levels of Nrf2 at 1 and 2 hours after treatment and a concomitant decrease in cytosolic Nrf2. It also increased hepatic thioredoxin, a protein whose gene transcription is induced by nuclear Nrf2. Levels of nuclear Nrf2 were at a plateau from 4 to 6 hours after T3. Rectal temperature of the animals rose from 36.6°C to 37.5°C as did liver O2 consumption. Serum and liver 8-isoprostanes levels increased (p < 0.05) from 38.4 ± 4.0 pg/mL (n = 4) to 69.2 ± 2.0 pg/mL (n = 3) and from 0.75 ± 0.09 ng/g liver (n = 3) to 1.53 ± 0.10 ng/g liver (n = 5), respectively. In the group of rats pretreated with NAC, the increase in cytosol-to-nuclear translocation of Nrf2 was only 28% that induced by T3. In addition, T3 induced liver Akt and p38 activation during the period of 1–4 hours after T3 administration. p38 activation at 2 hours after T3 administration was abolished in NAC-pretreated animals.
Conclusions:
In vivo T3 administration leads to a rapid and transient cytosol-to-nuclear translocation of liver Nrf2. This appears to be promoted by a redox-dependent mechanism as it is blocked by NAC. It may also be contributed by concomitant p38 activation, which in turn promoted Nrf2 phosphorylation. Nrf2 cytosol-to-nuclear translocation may represent a novel cytoprotective mechanism of T3 to limit free radical or electrophile toxicity, as this would likely entail promoting thioredoxin production.
Introduction
In addition to these cytoprotective mechanisms, induction of ROS and electrophiles secondarily upregulates antioxidant proteins and phase 2 detoxifying enzymes through activation of transcription factor nuclear transcription factor erythroid 2-related factor 2 (Nrf2) (13,14). Under physiological conditions, signaling via Nrf2 is impeded by the negative regulator Kelch-like ECH-associating protein 1 (Keap1), which acts both as a redox sensor through its cysteine-151 and as a subunit of E3 ubiquitin ligase, thus promoting continuous proteasomal degradation of Nrf2 (13 –15). Increased cellular ROS generation, in turn, promotes modification of Keap1-cysteine-151 to a Keap1-Cul3-Rbx1 E3 ubiquitin ligase complex form, which does not have anti-Nrf2 effects (15). This conversion is associated with increasing Nrf2 levels, its translocation into the nucleus, and its interaction with antioxidant response elements to activate target gene transcription (15). Nrf2 is sensitive to a rather low oxidative stress status (16) as that induced by the calorigenic action of T3 (4,6). The objective of this study was to test the hypothesis that in vivo T3 administration triggers cytosol-to-nuclear translocation (a putative activation step) of Nrf2 in rat liver by a redox-dependent mechanism, such as that which would be inhibited by the antioxidant N-acetylcysteine (NAC) (9). Here, we studied not only liver Nrf2 levels in cytosol and nucleus after administration of T3, with and without NAC pretreatment, but also hepatic levels of thioredoxin as a prototypical Nrf2-regulated protein (13) and activation of hepatic p38 and Akt (protein kinase B). Hepatic p38 and Akt are kinases that have been proposed to regulate the activity of Nrf2 by phosphorylation (15).
Materials and Methods
Animal treatments
Male Sprague-Dawley rats (Animal facility of the Institute of Biomedical Sciences, Faculty of Medicine, University of Chile) weighing 180–200 g were housed on a 12-hour light/dark cycle and were provided with rat chow and water ad libitum. Animals received a single intraperitoneal dose of 0.1 mg T3/kg body weight or equivalent volumes of hormone vehicle (0.1 N NaOH, controls) at time zero. Studies with NAC were carried out in the described groups receiving either 0.5 g/kg of NAC or saline, 0.5 hour before T3 administration. Therefore, there were four experimental groups: (i) controls, (ii) T3, (iii) NAC, and (iv) NAC + T3. Studies were performed for up to 48 hours after hormone treatment. T3-induced calorigenesis was assessed by the rectal temperature of the animals by means of a thermocouple (Cole-Palmer Instrument Company, Chicago, IL) and the rate of O2 consumption measured polarographically in liver perfusion experiments as previously described (3). Blood samples were obtained by cardiac puncture in rats anesthetized (1 mL/kg) with zolazepam chlorhydrate (25 mg/mL) and tiletamine chlorhydrate (25 mg/mL) ip (Zoletil 50; Virbac S/A, Carros, France) for measurement of serum 8-isoprostanes (ELISA; Cayman Chemical Co., Ann Arbor, MI), and liver samples were taken, frozen in liquid nitrogen, and kept at −80°C for measurement of 8-isoprostanes, Nrf2, p38, and Akt. Other studies were performed with the Nrf2 activator oltipraz (17). Rats were given either 150 mg/kg ip of oltipraz or corn oil vehicle (controls) per day for 4 consecutive days (LKT Laboratories, Inc., St. Paul, MN). Experimental animal protocols and animal procedures complied with the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, NIH Publication 86-23, revised 1985).
Western blot analysis of Nrf2, thioredoxin, p38, and Akt
Liver samples (100–500 mg) frozen in liquid nitrogen were homogenized and suspended in a buffer solution (pH 7.9) containing 10 mM HEPES, 1 mM EDTA, 0.6% Nonidet P-40, 150 mM NaCl, and protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 mM orthovanadate). Nuclear protein extracts (100 μg) and soluble protein fractions (60 μg) were separated on 12% polyacrylamide gels using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (18) and transferred onto nitrocellulose membranes (19), which were blocked for 1 hour at room temperature with Tris buffered saline (TBS) containing 5% bovine serum albumin. The blots were washed with TBS containing 0.1% Tween 20 and hybridized with either rabbit monoclonal antibodies for phospho-Akt, phospho-p38, and Akt (Cell Signaling Technology, Inc., New York, NY), rabbit polyclonal antibodies for Nrf2, thioredoxin, and p38 (Abcam, Cambridge, MA), or mouse monoclonal antibodies for β-actin (ICN Biomedicals, Inc., Aurora, OH) and lamin A/C (BD Transduction Laboratories, San José, CA). In all determinations, anti-β-actin was used as internal control for cytosolic fractions, whereas anti-lamin A/C was employed as internal control for nuclear fractions. After extensive washing, the antigen–antibody complexes were detected using horseradish peroxidase goat anti-rabbit IgG or goat anti-mouse IgG and a SuperSignal West Pico Chemiluminescence kit detection system (Pierce, Rockford, IL).
Statistics
Values shown correspond to the means ± SEM for the number of separate experiments indicated. One-way analysis of variance and the Newman–Keuls test assessed the statistical significance (p < 0.05) of differences between mean values. Statistical analysis of the studies with combined NAC and T3 treatments was carried out by two-way analysis of variance.
Results
There was a significant and progressive increase in the rectal temperature of rats given a single dose of 0.1 mg T3/kg when measured at 2–24 hours. This was followed by a decline at 36 hours, with the temperature stabilizing at 48 hours (Fig. 1A). Liver O2 consumption was increased at 2 hours compared with rats not given T3 and progressively increased from 2 to 24 hours (Fig. 1B). Under these conditions, T3 administration was associated with a 105% increase in the hepatic 8-isoprostanes (p < 0.05). This was suppressed by NAC pretreatment before T3 administration (control, 0.74 ± 0.09 ng/g liver [n = 3]; T3, 1.53 ± 0.10 [n = 5]; NAC, 0.87 ± 0.09 [n = 3]; NAC + T3, 0.86 ± 0.10 [n = 3]). There was a 15% increase in liver O2 consumption associated with T3 administration (p < 0.05). This increase was reduced by 67% (p < 0.05) in NAC-pretreated rats (control, 1.95 ± 0.05 μmol/(g liver·min) [n = 5]; T3, 2.25 ± 0.02 [n = 3]; NAC, 1.90 ± 0.04 [n = 3]; NAC + T3, 2.00 ± 0.05 [n = 3]).
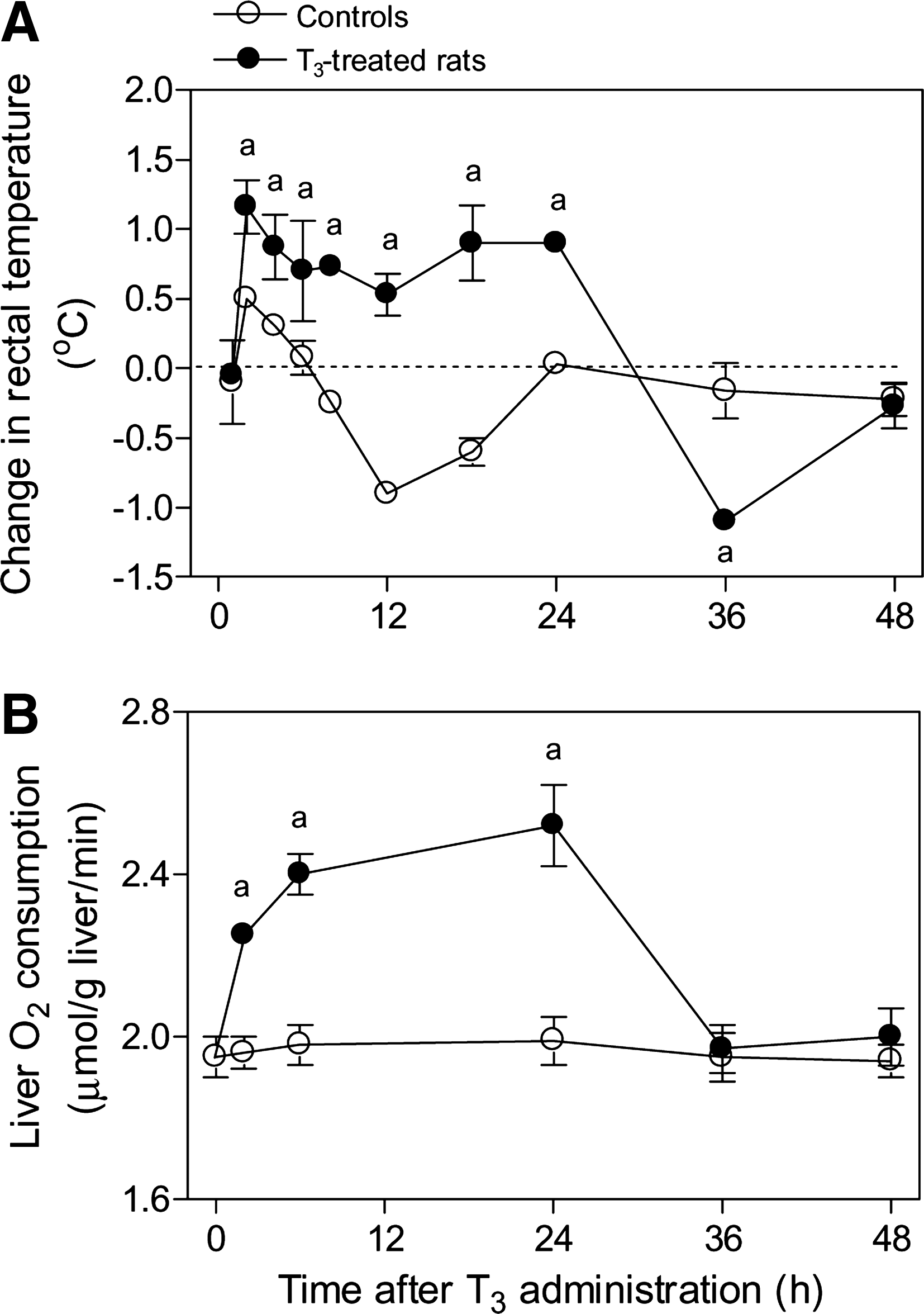
Time course study of the changes in the rectal temperature of the animals
Following T3 administration, there was a 50% decrease in hepatic cytosol Nrf2 at 1 hour and a 66% decrease at 2 hours (Fig. 2A). This was associated with a 99% increase in hepatic nuclear Nrf2 at 1 hour and a 188% increase in hepatic nuclear Nrf2 at 2 hours (Fig. 2B) (p < 0.05 for T3 group vs. control group). Both cytosolic and nuclear Nrf2 returned to control values at 4 and 6 hours after T3 administration (Fig. 2A, B). The change in the cytosol-to-nuclear ratio was taken here and elsewhere as being consistent with the activation of hepatic Nrf2. It occurred in a similar time frame as the development of oxidative stress. Thus, there were 105% and 55% increases (p < 0.05) in the hepatic levels of 8-isoprostanes observed at 2 and 6 hours, respectively, based on values in the control group, and there were 80%–90% increments in the serum levels of 8-isoprostanes (p < 0.05) at 1–6 hours after T3 treatment (Fig. 3).
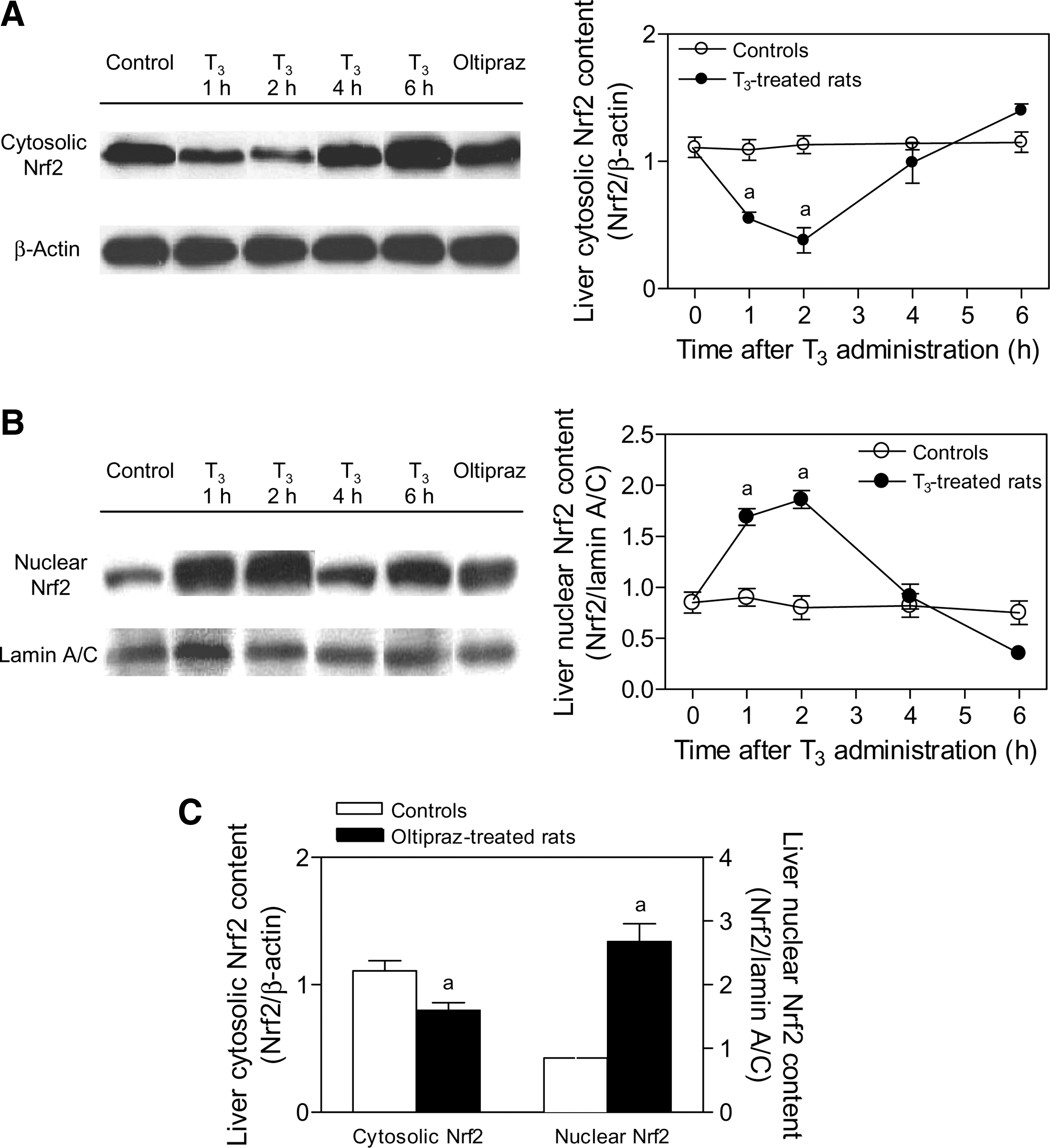
Time course study of the effects of T3 administration on liver nuclear transcription factor erythroid 2-related factor 2 (Nrf2) protein expression in rats.
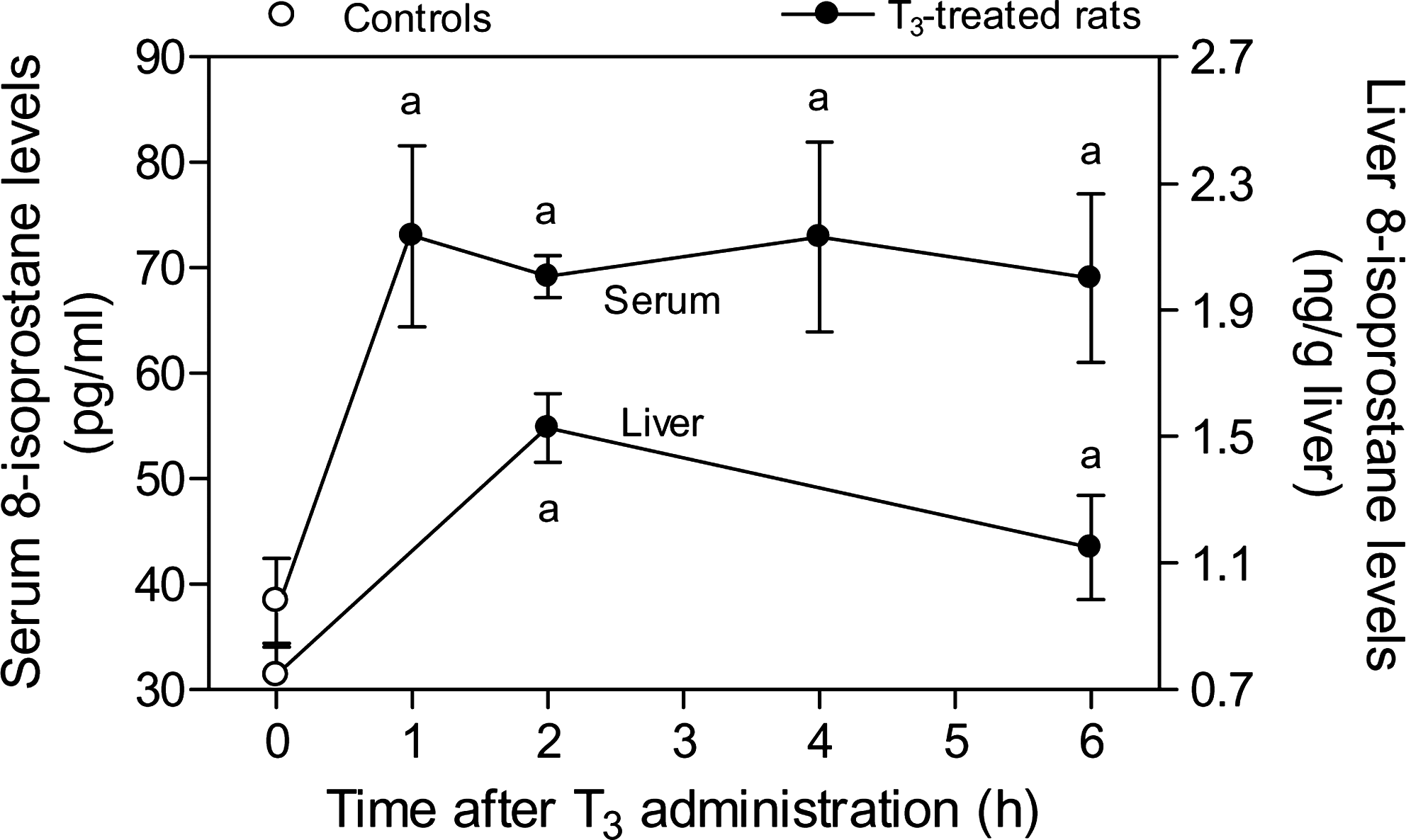
Time course study of the effects of T3 administration on hepatic and serum 8-isoprostanes levels in rats. Data are means ± SEM (n = 3–15); a p < 0.05, compared with control values at time zero.
The methodology for discernment of cytosol and nuclear Nrf2 was evaluated by testing the effects of oltipraz administration, a known activator of Nrf2 and its controlled genes (17,20). Oltipraz administration was associated with a 28% reduction in cytosolic Nrf2 levels and 215% increase in nuclear Nrf2 content when compared with values in control rats (p < 0.05) (Fig. 2C). Similarly to T3 administration (Fig. 3), oltipraz treatment enhanced the serum levels of 8-isoprostanes, an effect that was of a higher magnitude than T3 (controls, 28 ± 4 pg/mL [n = 15]; oltipraz-treated rats, 203 ± 37 [n = 4]; 4.3-fold increase; p < 0.05 by Student's t-test for unpaired data).
Rats (NAC + T3) given 0.1 mg/kg T3 and 0.5 g/kg NAC before T3 administration (Fig. 2A, B) had less of a decrease in cytosol Nrf2 and less of an increase in nuclear Nrf2 than rats given T3 alone (Fig. 4A, B). At 2 hours after T3 treatment, the decrease in cytosolic Nrf2 in NAC-pretreated rats was 9% of that in animals given T3 alone (Fig. 4A) and the increase in nuclear Nrf2 was 28% (Fig. 4B), respectively. Further, evaluation of Nrf2, which targets thioredoxin (20), revealed 220% enhancement (p < 0.05) in its protein expression at 2 hours after T3 administration, compared with control values (Fig. 5).
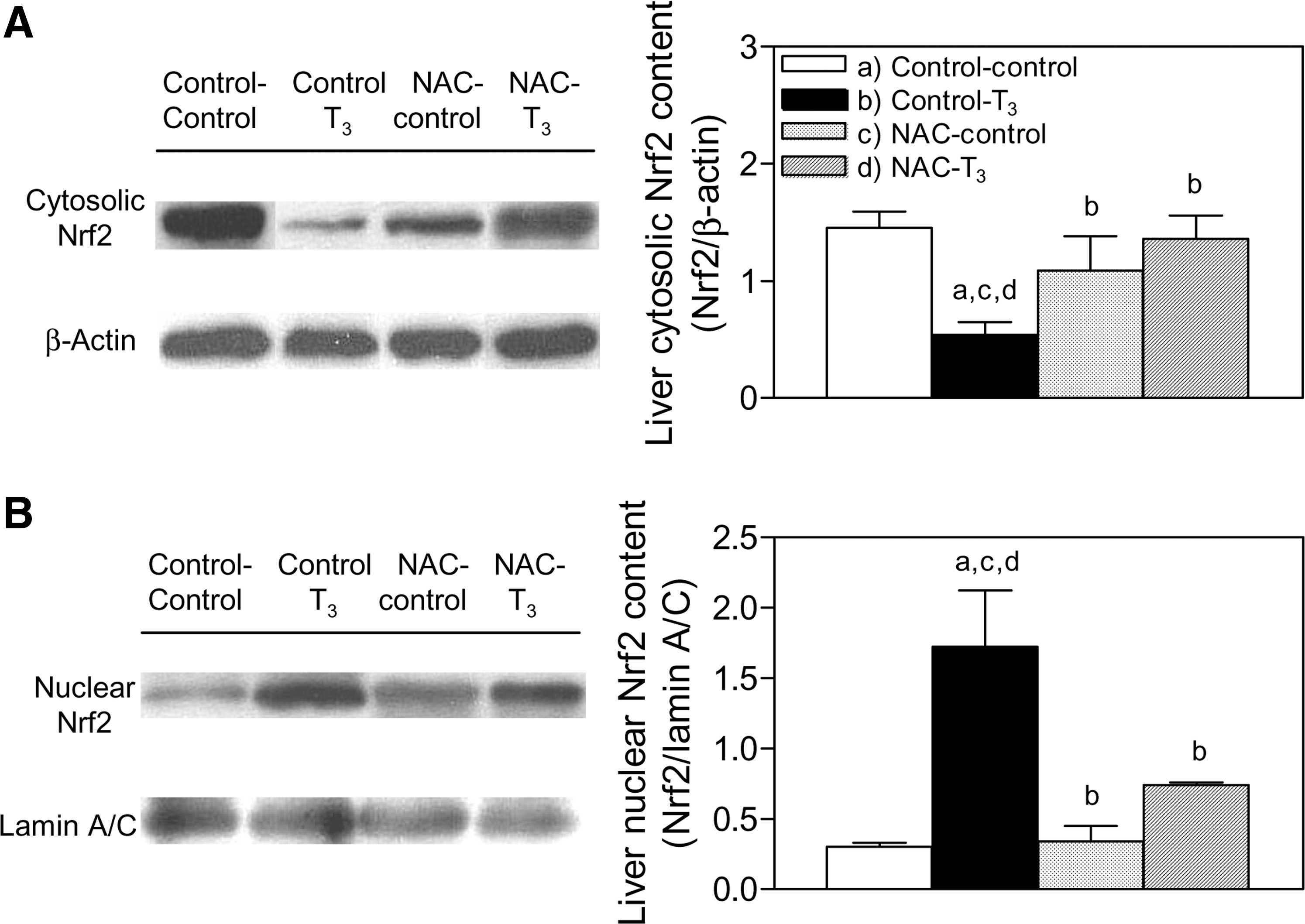
Effect of N-acetylcysteine (NAC) pretreatment on liver Nrf2 protein expression induced by T3 administration in rats. Determinations were carried out at 2 hours after T3 treatment.
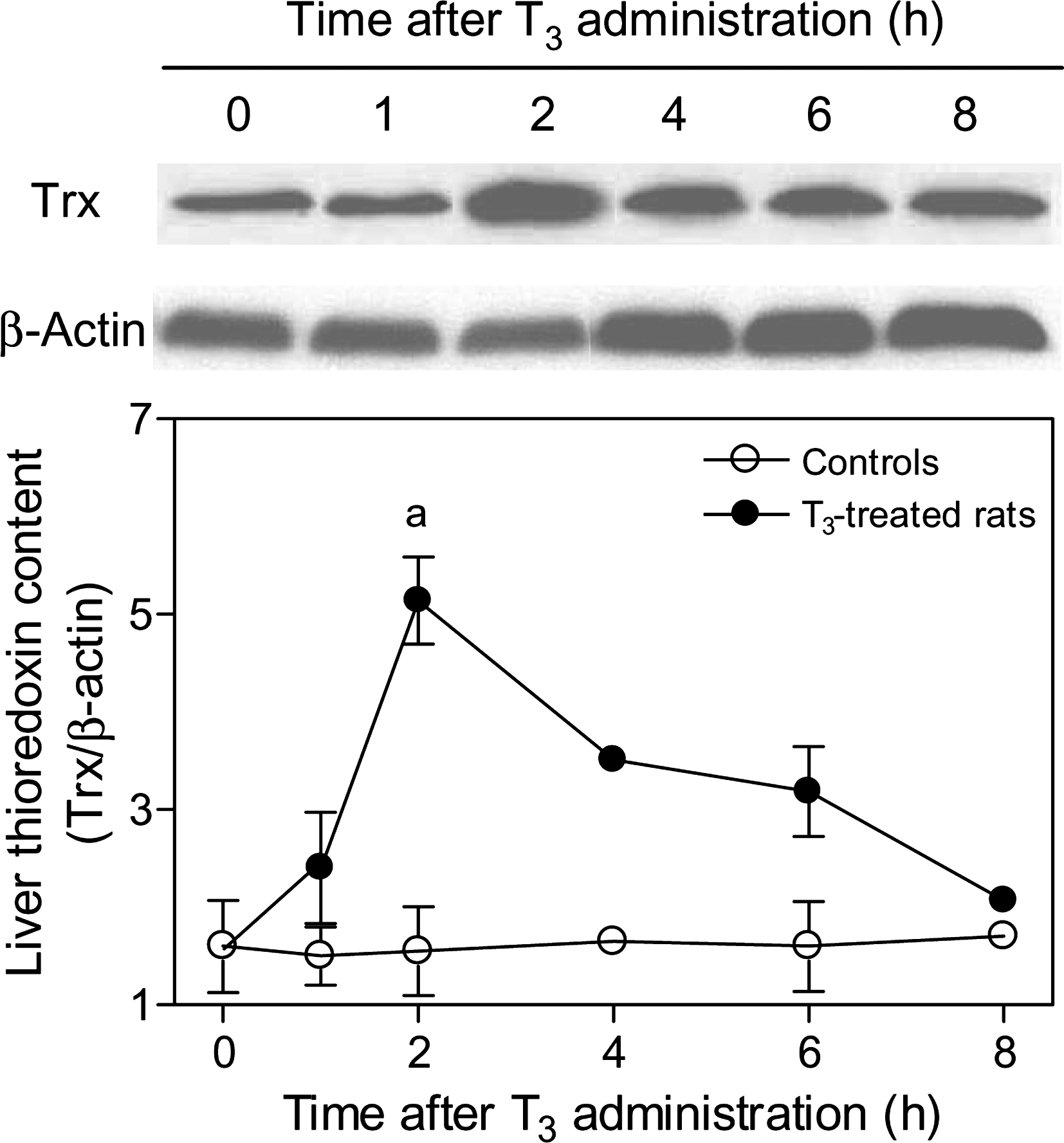
Time course study of the effects of T3 administration on liver thioredoxin (Trx) protein expression in rats. Representative blots of cytosolic thioredoxin (12 kDa) and β-actin (43 kDa) expression using 50 μg of soluble protein from a different rat of each group studied (upper panels). The lower panel shows the respective densitometric quantification expressed as thioredoxin/β-actin ratios to compare lane-to-lane equivalence in total protein content (lower panel). Values shown are means ± SEM (n = 3–5); a p < 0.05, compared with the respective control values.
The administration of 0.1 mg/kg T3 was associated with 180% increase (p < 0.05) in the phosphorylated Akt/nonphosphorylated Akt (Akt-OP/Akt-OH) ratios (Fig. 6A). The phosphorylated p38/nonphosphorylated p38 (p38-OP/p38-OH) ratios were increased by 498%, 221%, and 175% (p < 0.05) at 1, 2, and 4 hours after T3 treatment, respectively (Fig. 6B). At 2 hours after T3 administration, Akt-OP/Akt-OH ratios were enhanced by 180% over control values and by 113% in NAC-pretreated T3-treated rats over NAC control animals, leading to a net NAC-dependent nonsignificant 28% diminution in T3-induced Akt activation, in the absence of significant effects by NAC itself (Fig. 7A). Further, p38-OP/p38-OH ratios were increased by 222% by T3 at 2 hours after treatment, compared with controls (p < 0.05), an effect that was suppressed by NAC pretreatment prior to T3. Under these conditions, NAC did not modify liver p38 activation as evidenced by the lack of effects on the hepatic p38-OP/p38-OH ratios when given alone to euthyroid rats (Fig. 7B).

Time course study of the effects of T3 administration on liver Akt and p38 phosphorylation in rats. Representative blots of liver soluble protein fractions (60 μg) for detection of nonphosphorylated Akt (Akt-OH; 60 kDa)
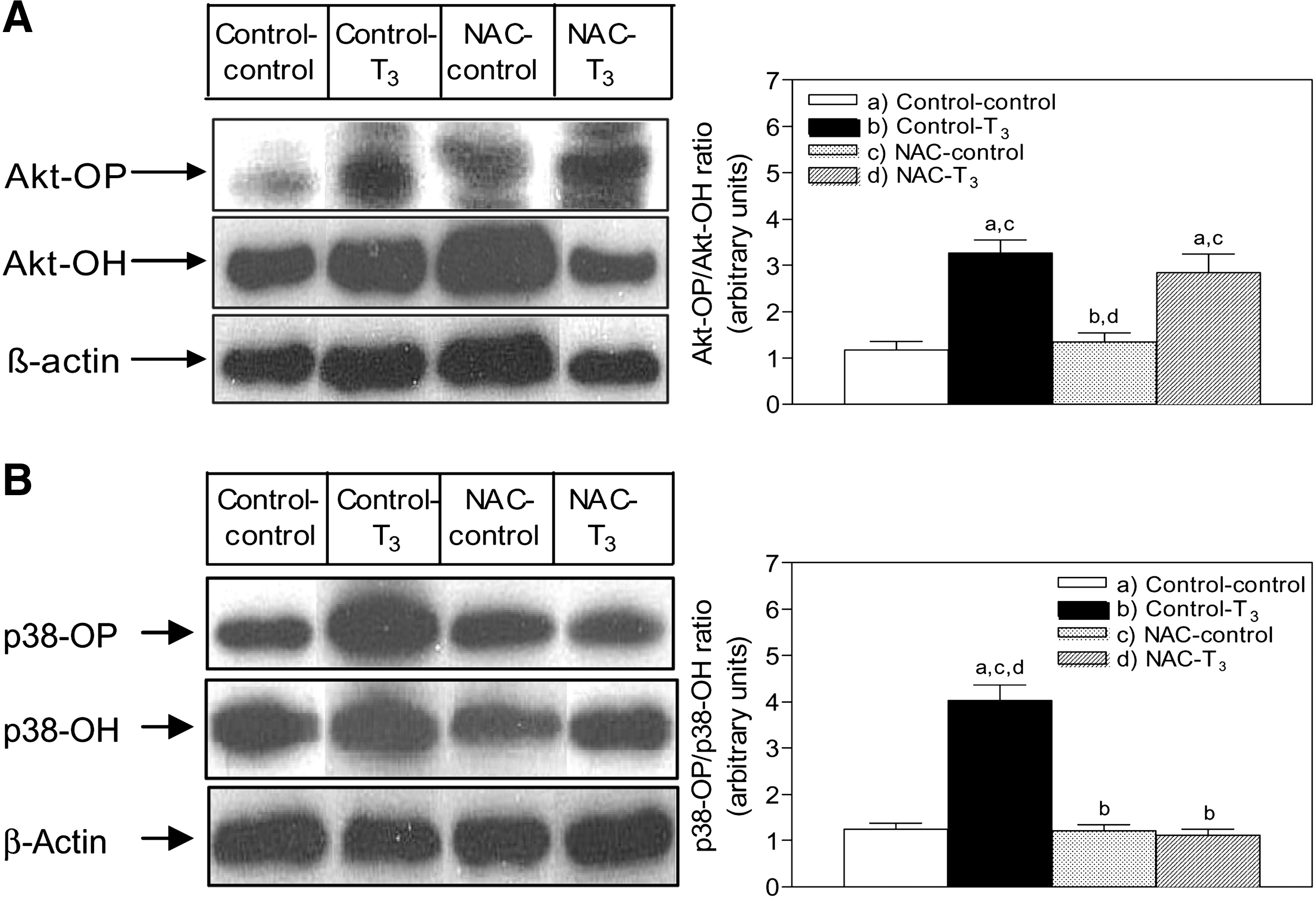
Effect of NAC pretreatment on liver Akt and p38 phosphorylation induced by T3 administration in rats. Determinations were carried out at 2 hours after T3 treatment.
Discussion
Previous studies have shown that induction of genes involved in cytoprotection occurs after the administration of T3 to rats at doses that have a calorigenic effect (6,8). T3 induced a biphasic response in rectal temperature probably related to upregulation of the expression of respiratory genes through genomic mechanisms (4,6). The late decline in rectal temperature at 36 hours after T3 treatment was probably mediated, at least in part, by a negative feedback of T3 on TSH (21). In the present study, we have found that administration of T3 at calorigenic doses, involving higher rates of liver O2 consumption, was associated with apparent translocation of the cytoprotective transcription factor Nrf2 from the cytosol to the nucleus, consistent with activation of Nrf2. The enhancement in the hepatic nuclear abundance of Nrf2 with diminution in that of cytosolic Nrf2 was an early event occurring at 1–2 hours after T3 treatment, similar to most rapid nongenomic actions of thyroid hormones (22 –25). This is the time period during which there is a development of oxidative stress as evidenced by the enhanced hepatic and serum levels of 8-isoprostanes. Interestingly, one of the target genes for Nrf2, that is, the thioredoxin gene, also appears to be activated, as thioredoxin levels are increased at 2 hours after T3 administration. Thioredoxin is a major component of the cellular antioxidant system (14,15). The major finding of the present study was that NAC pretreatment markedly decreased the T3-induced activation (cytosol-to-nuclear translocation) of liver Nrf2. It also suppressed the T3 induction of increases in liver 8-isoprostane levels and O2 consumption. In the present study, we did not measure serum NAC levels. In prior studies under similar experimental conditions, however, the administration of 0.5 g/kg of NAC to rats led to a rapid increase of the serum NAC levels at 0.5 hour and a fall to undetectable concentrations after 48 hours (9). We postulate that the increase in the oxidative stress status of the liver associated with T3-induced calorigenesis is an important factor for Nrf2 upregulation, which may expand the pool of free Nrf2. This can be achieved by Keap1 inactivation associated with T3-induced ROS promoting Keap1-cysteine-151 oxidation, which inhibits its anti-Nrf2 effects (14,15,26).
In addition to the ROS-induced Keap1-mediated ubiquitination/degradation mechanism controlling Nrf2 activity, subcellular localization and posttranslational modification of Nrf2 are also involved (15,26). In the latter case, it has been proposed that Nrf2 phosphorylation plays a role in Nrf2 activation either (i) by increasing free Nrf2 levels, as phosphorylated Nrf2 is no longer a substrate for the Keap1-Cul3-Rbx1 E3 ubiquitin ligase complex (15,27), and/or (ii) by enhancement of the binding affinity of Nrf2 to antioxidant response elements (15). In this respect, data presented suggest that Nrf2 phosphorylation by p38, rather than Akt, may play a role in Nrf2 upregulation by T3 administration, considering (i) that mitogen-activated protein kinase (MAPK) p38 is concomitantly activated as shown by the significant increase in the hepatic p38-OP/p39-OH ratio over control values, and (ii) that both Nrf2 and p38 activation by T3 are suppressed by NAC treatment prior to T3. Activation of p38 consists of the initial stimulation of an MAPK kinase kinase by ROS, with the sequential phosphorylation of an MAPK kinase and p38 (28). Although p38 phosphorylation coincides with Nrf2 activation by T3, recent studies revealed that phosphorylation of Nrf2 by MAPKs including p38 has a limited contribution in modulating Nrf2 activity and suggested that indirect mechanisms at the translational level may be involved through control of Nrf2 protein synthesis by MAPKs (29). This contention is supported by the interaction of thyroid hormones with a plasma membrane receptor on integrin αVβ3 to activate MAPK signaling, the affinity of the receptor for T3 being lower than that for thyroxine (22).
Although T3-induced redox upregulation of liver Nrf2 activity and content is a rapid event occurring within 1–2 hours after T3 administration, it returned toward basal levels from 4 hours onward. Surprisingly, downregulation of hepatic Nrf2 after T3-induced activation is observed under conditions in which a pro-oxidant state is present, as evidenced by the calorigenic response lasting for 24 hours and involving higher serum and hepatic 8-isoprostanes levels, an indicator of free radical-dependent lipid peroxidation. Liver Nrf2 downregulation under sustained oxidative stress conditions induced by T3 may be ascribed to the enhancement in the expression of Nrf2 inhibitor Keap1. This proposal is based on data concerning the role of Nrf2 in cancer promotion, showing that enhanced Nrf2 activity and Nrf2-dependent gene expression in tumor cells is related to dysfunctional Keap1-Nrf2 interaction and/or Keap1 downregulation by promoter hypermethylation (30 –32). Conversely, T3-induced Nrf2 downregulation after activation may be explained in terms of Keap1 upregulation due to hypomethylation of the Keap1 gene, a feature that is required for active gene transcription (33). This contention is supported by the finding that T3 administration results in the demethylation of a specific DNA cytosine in the 3' region of the S14 gene, which is associated with increased expression of tissue-specific genes (34). Recently, an autoregulatory loop between Keap1 and the ubiquitination-related proteins Cul3 and Rbx1 controlling Nrf2 cellular abundance has been established, with Nrf2 activation upregulating the expression of Keap1, Cul3, and Rbx1, which in turn increase Nrf2 degradation (35,36). The influence of T3 on liver Keap1, Cul3, and Rbx1 expression is under investigation in our laboratory.
In summary, data presented show that T3 administration to rats leads to a rapid and transient increase in liver Nrf2 activation, as evidenced by the lower cytosolic and higher nuclear Nrf2 protein levels observed. Liver Nrf2 activation induced by T3 appears to be a redox-dependent process, which may be contributed by Nrf2 phosphorylation related to p38 activation. This would represent an alternate cytoprotective mechanism of T3 action against free-radical or electrophile toxicity, in addition to that afforded by NF-κB, STAT3, and/or AP-1 upregulation (6).
Footnotes
Acknowledgment
This work was supported by a grant (no. 1090020) from FONDECYT (Chile).
Disclosure Statement
The authors declare that no competing financial interests exist.