
Abstract
Background:
Superagonist analogs of human thyroid-stimulating hormone (hTSH) may stimulate the uptake of 131I-iodide and 18F-fluorodeoxyglucose (18F-FDG) in thyroid carcinomas to a greater degree than hTSH. We herein report the potency and efficacy of two hTSH analogs, TR1401 and TR1402, to stimulate radioiodide and 18F-FDG uptake in FRTL-5 cells and compared the effects of hTSH and TR1401 on radioiodide uptake in the thyroid in vivo in mice.
Methods:
The effects of hTSH analogs on intracellular levels of cAMP, uptake of 131I-iodide, and 18F-FDG were studied in FRTL-5 cells to determine the stimulatory potency and efficacy of the compounds by calculating half-maximum effective concentration (EC50) values and maximal stimulatory effects (Emax). Biodistribution studies (n = 96) and positron emission tomography/computed tomography imaging studies (single animals) on thyroid 125I/124I-iodide uptake were performed with T3-suppressed CD-1 mice in a dose-dependent manner (3, 10, and 30 μg/animal).
Results:
The EC50 values of TR1401 and TR1402 demonstrated a 90-fold or 800-fold higher potency for their capacity to increase intracellular cAMP levels in comparison with hTSH (p < 0.05). Similar results were demonstrated for the stimulation of 18F-FDG uptake. Bovine TSH, TR1401, and TR1402 were 85%–490% more potent to increase iodide uptake than hTSH (p < 0.05). TR1402 was 30% more efficacious to stimulate iodide uptake than hTSH. The agonist-induced increase in radiotracer uptake was paralleled by increases in NIS and GLUT-1 expression. Ex vivo biodistribution studies showed an increased iodide uptake in the thyroid of TR1401-treated mice at the low dose of 3 μg/animal in comparison with hTSH-treated mice (n = 16, p < 0.05). Positron emission tomography/computed tomography imaging studies confirmed the increased thyroidal iodide uptake in TR1401-treated mice in vivo.
Conclusions:
TR1401 and TR1402 have considerably higher potency than hTSH to stimulate thyroidal iodide and 18F-FDG uptake in vitro. Moreover, in vivo studies indicated that at low but not higher doses, TR1401 induced an enhanced ability for the thyroid to concentrate iodide compared with hTSH. These properties makes TR1401 and TR1402 interesting candidates for use in humans to enhance uptake of radioiodine and 18F-FDG by metastases and recurrences of thyroid carcinoma.
Introduction
The success of radioiodine therapy, as well as the diagnostic sensitivity of radioiodine scintigraphy and 18 F-FDG-PET, is directly related to the amount of tracer accumulating in the target lesions. TSH agonists with higher potency and efficacy than hTSH could therefore help improve the above-mentioned diagnostic and possible future approved therapeutic procedures. In comparison with hTSH, bovine TSH (bTSH) is known to display high affinity and superagonistic cAMP activity at the hTSH receptor (TSHR), owing to the presence of positively charged lysine residues in the amino acid sequence of the surface-exposed loops of the α subunit (16). Most likely, the high bioactivity of bTSH is attributed to the interaction of the lysine residues with the negatively charged Glu297 and Asp382 of the TSHR (17).
bTSH has both acute allergic and immunogenic side effects in humans; thus, it is not used in the clinical setting. Therefore, we have developed analogs of hTSH by using defined design strategies (16,18 –20) to obtain improved TSH receptor binding affinity as well as increased signal transduction (21,22). The hTSH analog TR1401, whose sequence differs from hTSH by four additional positively charged amino acids that are also present in bTSH, was characterized as a superagonistic hormone by cAMP assays (16,20,23). This strategy for the design of superagonists for the TSHR may lead to compounds with markedly increased therapeutic potential and provide a useful tool to characterize extrathyroidal TSH receptor systems and to increase our knowledge on their potential physiological role (24,25).
In this work, we studied the potency and efficacy of TR1401 and TR1402, two closely related analogs of hTSH that both differ from hTSH by four additional positively charged amino acids located in the α-L1 loop of the glycoprotein hormone (Fig. 1). The outcome variable of this investigation was the ability of these compounds to stimulate uptake of radioiodide and 18 F-FDG in the rat thyroid cell line FRTL-5 as an in vitro model of well-differentiated thyroid tissue (26 –29). As a proof of concept, the effects of hTSH and TR1401 on incorporation of radioiodide in the thyroid in vivo were analyzed by biodistribution and PET/computed tomography (CT) studies.
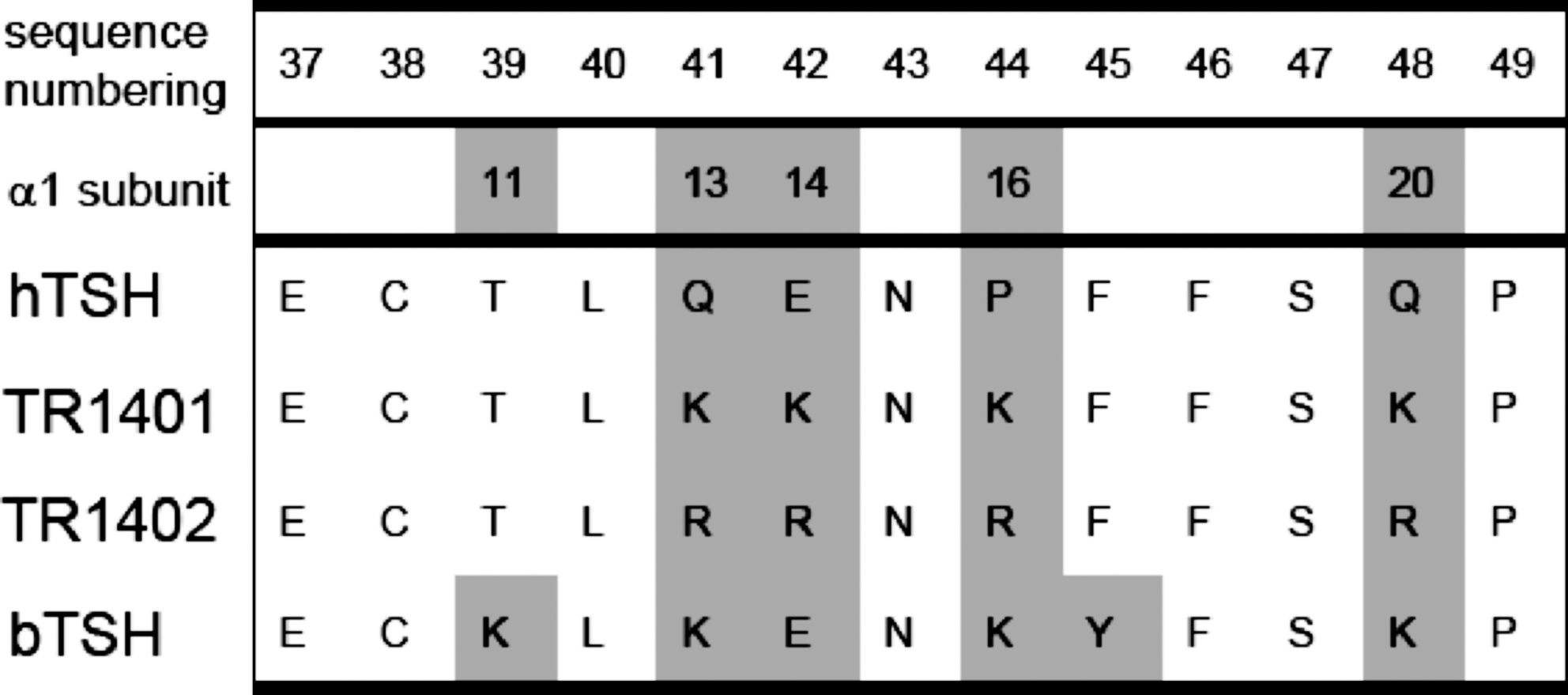
Amino acid sequence differences between hTSH and the human TSH analogs TR1401 and TR1402. For comparison, the sequence of bTSH is displayed. Sequence numbering in this alignment is shown according to bovine α subunit sequence with signal peptide (1st row) (23). Differences in the amino acid sequences are highlighted and the sequence numbering is shown in the 2nd row according to human α subunit sequence without signal peptide. TR1401 and TR1402 differ by lysine-to-arginine replacement in positions 13, 14, 16, and 20 of the α subunit that plays a major role for the TSH receptor-mediated agonist activity. hTSH, human thyroid-stimulating hormone; bTSH, bovine TSH.
Materials and Methods
General
Na131I (product code IBSSO) was obtained from Perkin Elmer (Rodgau-Jügesheim, Germany). 18 F-FDG was purchased from PET Net GmbH (Erlangen, Germany). Bovine TSH (bTSH), LY294002, H89, Kaighn`s modified Ham's F-12, Nutrient Mixture F-12 Coons modification medium, insulin, hydrocortisone, transferrin, Gly-His-Lys acetate salt, somatostatin, L-glutamine, G418, and 3-isobutyl-1-methyl-xanthine (IBMX) were supplied by Sigma-Aldrich. Dulbecco's modified Eagle's medium (DMEM), Hank's buffered saline solution (HBSS), fetal calf serum (FCS), phosphate buffered saline (PBS), and trypsin/EDTA were obtained from Invitrogen/Gibco (Germany). Fetal bovine serum (FBS) was purchased from GlobalStem (Rockville, MD). Cyclic AMP EIA kit was purchased from Cayman Chemical (Ann Arbor, MI). FRTL-5 cells were generously provided by Dr. L.D. Kohn (Interthyr Research Foundation, Baltimore, MD) or obtained from the European Collection of Cell Cultures (no. 91030711). Solutions of highly purified bTSH, hTSH, TR1401, or TR1402 preparations were quantified for mass content by HPLC as described prevously (16), prior their use for stimulation experiments.
Superagonist TSH analogs: TR1401 and TR1402
The human TSH analogs TR1401 and TR1402 were produced at Trophogen, Inc. (Rockville, MD), following the procedures for site-directed mutagenesis and expression of recombinant hormones as decribed previously (16,20). Both, TR1401 and TR1402 were produced in stably transfected Chinese hamster ovary (CHO) cells, purified by a combination of dye, ion exchange, and gel filtration HPLC. TR1401 is a highly purified recombinant human TSH analog with four substitutions (Q13K+E14K+P16K+Q20K, numbering without signal peptide, Fig. 1) in the α subunit of the glycoprotein. TR1402 represents the arginine analog with the corresponding four substitutions in the α subunit (Q13R+E14R+P16R+Q20R, numbering without signal peptide, Fig. 1). The final TR1401 and TR1402 preparations were more than 98% pure based on SDS-PAGE and reverse-phase HPLC.
Cell culture
The rat thyroid cell line FRTL-5 was grown in Kaighn`s F12 medium supplemented with a six-hormone mixture [10 mg/L insulin, 5 mg/L transferrin, 10 μg/L somatostatin, 10 nM hydrocortisone, 10 μg/L glycyl-L-histidyl-L-lysine acetate, and 1 U/L bovine TSH; 6H medium (30)] and 5% FCS at an atmosphere of 5% CO2 at 37°C. FRTL-5 cells were routinely subcultured every 3–4 days and the cell viability was proven by trypan blue staining.
cAMP assay
The effect of TSH analogs on intracellular cAMP levels was determined by a competitive enzyme immunoassay as described previously (31). In brief, FRTL-5 cells were grown in Coon's modified F-12 medium supplemented with 5% FBS and a 6-hormone mixture (6H medium) including bTSH for 3 days. Cells were plated in 96-well plates and grown in 6H medium without bTSH (5H medium) for 6 days. The FRTL-5 cells were incubated with different concentrations of either hTSH (Thyrogen), bTSH, TR1401, or TR1402 preparations, diluted in medium supplemented with 1 mM IBMX, for 2 hours at 37°C. The medium was then collected and cAMP concentration was measured by a cyclic AMP EIA kit (Cayman Chemical, Ann Arbor, MI).
Determination of intracellular 131I-iodide or 18 F-FDG uptake
Following the procedure described previously (27), FRTL-5 cells (150.000/mL in 6H medium) were seeded in 12-multiwell culture plates and the medium was changed to a 5H medium (6H medium without bTSH) after 24 hours. TSH deprivation was continued for 5 days. Then, the medium was changed to fresh 5H medium (1.0 mL) and cells were treated with 2.3 pg/mL–2400 ng/mL hTSH, bTSH, TR1401 or TR1402, respectively. For inhibition experiments cells were pretreated with H89 (25 μM, selective inhibitor of PKA) or LY294002 (10 μM, selective inhibitor of PI3K) 30 minutes before activation with the TSH analogs. Two days after treatment, 18 F-FDG (0.1 MBq, 10 μL) was applied to each culture well and incubation was continued for 1 hour at 37°C or the medium was discarded and cells were washed with HBSS (containing 5.55 mmol/L glucose and 10 mmol/L HEPES) and a solution containing radioiodide (HBSS containing 10 μmol/L sodium iodide and 0.75–1.00 μCi/mL sodium 131I-iodide; 1.0 mL per well) was added and incubated for 10 minutes. The radiotracer uptake was terminated by cooling the cell layer with ice and an aliquot from the supernatant (50 μL) was withdrawn for radioactivity measurements. Each well was rapidly rinsed once with cold HBSS/HEPES. The cell layer was dissolved using a warm solution of sodium hydroxide (0.1 mol/L; 0.5 mL per well), and radioactivity measurements were performed with a γ-counter (Wallac Wizard, Perkin Elmer). After determination of protein concentration by the method of Bradford (32), the radiotracer uptake was expressed as percent of total radioactivity divided by protein mass (%/mg) and related to untreated cells.
Membrane preparation
Total cellular membranes were isolated from FRTL-5 cells that were cultured and treated as described for the uptake experiments. After stimulation with hTSH, bTSH, TR1401, or TR1402 (2.3 pg/mL − 2400 ng/mL), the membrane preparations were obtained by using a membrane protein extraction kit (ProteoExtract®, Merck Biosciences) according to the manufacturer's instructions. In some cases, the protein concentration of the final sample was increased by ultrafiltration using Roti-Spin Mini-10 inserts (Roth GmbH, Karlsruhe) and centrifugation at 14,000g for 15 minutes at 4°C. The protein concentration of each sample was determined by the QuantiPro™ BCA assay kit (Sigma) using bovine serum albumin (BSA) as a standard.
Western blot
The electrophoresis of samples from membrane preparations (each sample corresponding to 6.2 μg as determined by BCA assay using bovine serum albumin [BSA] as standard) was performed on precast NuPAGE® Novex Bis-Tris gel (Invitrogen) and transferred to nitrocellulose membranes by using a Hoefer miniVE blot module (Amersham Biosciences). SeeBlue® prestained standard and Magic Marker® (Invitrogen) were loaded on each gel as molecular weight standards. Nitrocellulose membranes were incubated in blocking solution (5% nonfat dry powdered milk in TBS buffer [50 mM Tris, 120 mM NaCl, pH 7.5]) for 2 hours at room temperature. A rabbit polyclonal anti-human glucose transporter-1 antibody (FabGennix Inc., 1:1000) or a polyclonal rabbit antibody to rat Na/I-Symporter (Acris EUD4101, 1:1000) and a rabbit anti-β-actin antibody (Sigma, 1:200) were used for primary antibody incubation overnight at 4°C. After washing in TBS buffer (0.1% Tween-20, 3 × 15 minutes), the membranes were incubated with an anti-rabbit secondary antibody coupled to horseradish peroxidase (Calbiochem, 1:1000) in TBS buffer for 1 hour at room temperature. After repeated washing (3 × 15 minutes) in TBS buffer (0.1% Tween-20), a chemoluminescence reagent (Amersham ECL Plus; GE Healthcare) was used for observation of the blotting result by a Fluor-S™ MultiImager using the software Quantity One® (vers. 4.3.0, Bio-Rad).
Statistics
Statistical analysis was performed using SPSS Software (version 17.0, SPSS Software GmbH). Only stimulation experiments that were perfomed within four “cell batches” (one “cell batch” was defined as cells obtained between 5 and 10 cell passage cycles) were analyzed to obtain mean values of the half-maximum effective concentration (EC50) for each TSH analog. This was necessary because the interassay deviation was found to be >80% when totally different cell batches of FRTL-5 cells were used for independent stimulation experiments with the same stimulant. This effect is possibly due to the well-known deviation of cell properties of FRTL-5 cells during cell culture (26). The curve fitting was analyzed using the software GraphPad Prism (GraphPad Software Inc., La Jolla, CA) and the equation for dose–response curve with variable slope. Within this data set, only experiments with adequate curve parameters were qualified for the calculation of mean values for the maximal efficacy or mean values of EC50 values. These curve parameters included (a) “goodness of fit” with R > 0.85, (b) SD (max. efficacy) < 15%, (c) dependency of top of the curve, LogEC50, and Hill slope: each below 0.5. All data are expressed as mean ± SEM or ± SD as indicated in the figure legends. For inhibition experiments, the control values were set to 100% to allow for pooling of data from individual experiments. All statistical results are based on the nonparametric Mann-Whitney test. The number of samples (n) used for statistical analysis and the number of independent experiments are given in the figure legends. Probability values (p) of 0.05 or less were considered to be significant.
Animals
All animal experiments were performed in compliance with the protocols approved by the Johns Hopkins University ACUC guidelines. Female CD-1 mice (25–35 g) were purchased from Charles River Laboratories (Wilmington, MA) and housed five per cage throughout the experiment. They were given food and water ad libitum. The mice were injected IP twice per day for 4.5 days in 12 hour intervals (7 AM and 7 PM) with 200 μL of T3 solution (15 μg/mL).
Ex vivo biodistribution
In the evening of day 5, 10 μg each of test compound (PBS alone, TSH or TR1401) in PBS (200 μL) was intraperitoneally (IP) injected. At 8 AM the following morning (13 hour post-treatment), [125I]NaI (2.3 μCi in 200 μL of sterile saline) was injected IP followed by sacrifice via cervical dislocation at 1, 4, 8, 12, 36, and 48 hour postradioiodide administration. The mice were immediately dissected and their thyroids removed along with the attached section of trachea. The tracheas were weighed and counted in a Wallac 4220 automated gamma counter. Percent injected dose per organ (% ID/organ) values were calculated by comparison of samples with a standard dose. One-way ANOVA t-tests were conducted to determine data significance between hTSH and TR1401 values. In addition, the one-parameter curve fitting of the time–activity curves for determination of the maximum uptake value was performed by nonlinear regression using GraphPad Prism software (one-phase exponential association equation; GraphPad Software Inc., La Jolla, CA). Differences of time–activity curves were tested by the extra sum-of-squares F-test and two-tailed p-values were calculated for the best-fit values of maximum uptake.
PET/CT imaging
Five T3-treated mice were selected to undergo PET-CT imaging and quantitation. Each mouse received PBS alone, 3 μg of TSH, 3 μg of TR1401, 10 μg of TSH, or 10 μg of TR1401 in the evening of day 5 after T3 suppression. Twelve hours after receiving the TSH analogs, the mice were injected IP with 254 μCi of [124I]NaI. The mice were then imaged using a GE eXplore Vista small animal PET scanner using a 10-minute acquisition in the 400–700 keV energy window. Images were reconstructed using a 2D OSEM algorithm. CT data were acquired using a Gamma Medica X-SPECT scanner and data were reconstructed using the manufacturer's software. Images were coregistered and displayed using AMIDE freeware (
Results
The TSH receptor activation in FRTL-5 cells by increasing concentrations of TSH analogs led to intracellular accumulation of cAMP with superior half-maximally effective concentration values (EC50) for TR1401 (31.4 ng/mL) and TR1402 (3.5 ng/mL) when compared with hTSH (2800 ng/mL) and bTSH (58.6 ng/mL) (Fig. 2). Thus, TR1401 was shown to be 90-fold more potent than Thyrogen in cAMP stimulation and this effect was even enhanced by TR1402, indicating a 800-fold increase in the potency to stimulate cAMP in FRTL-5 cells.
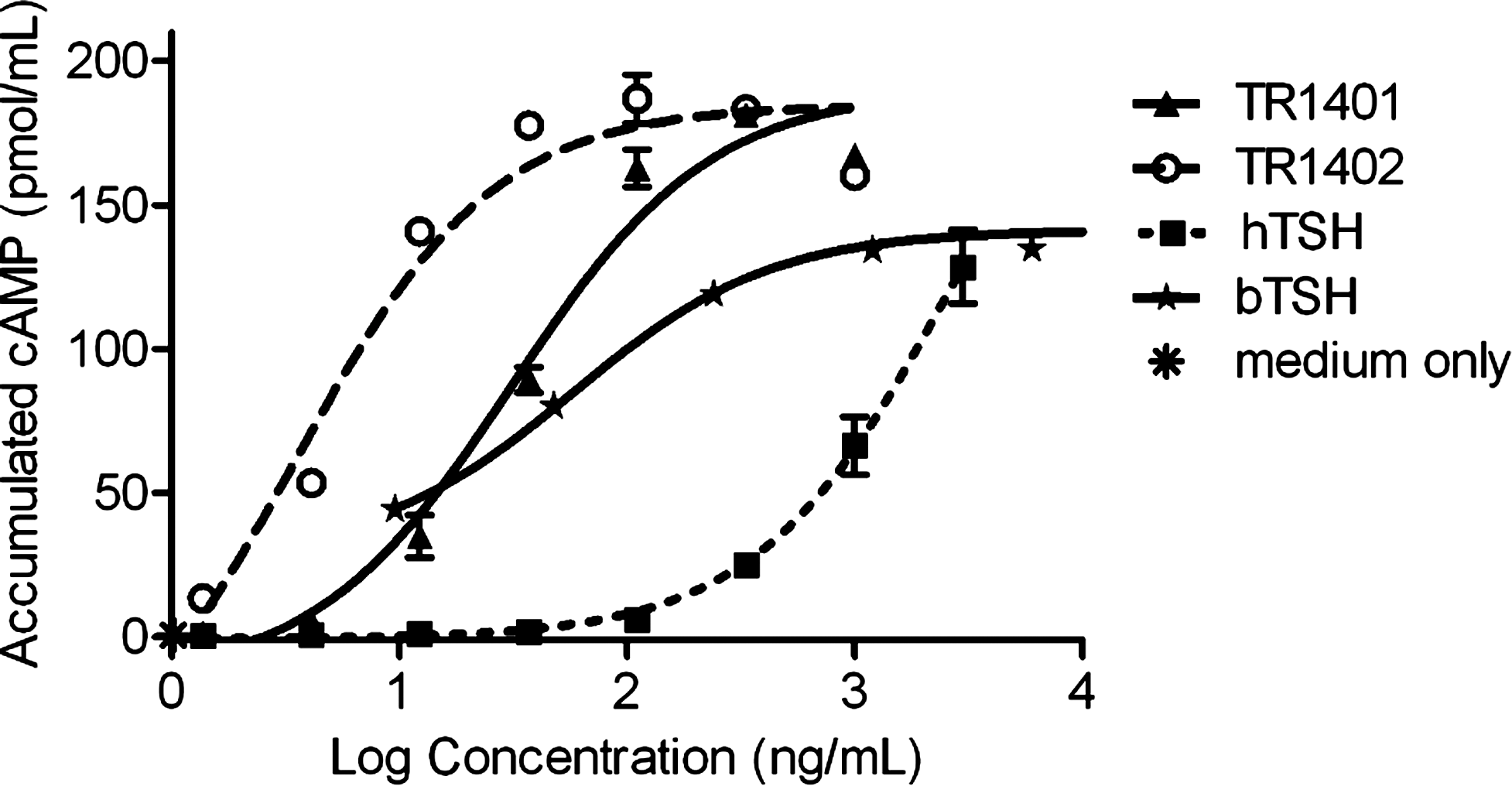
Cyclic AMP stimulation in FRTL-5 cells by hTSH (Thyrogen), bTSH, and TSH analogs TR1401 and TR1402. After being maintained for 6 days in the absence of TSH, the cells were incubated with indicated concentrations of hTSH, bTSH, TR1401, and TR1402 preparations for 2 hours at 37°C as described in Materials and Methods. The amount of cAMP accumulated in the medium was assayed by a cAMP EIA kit. Values are the mean ± SEM of triplicate determinations from a representative experiment, repeated three times.
We studied the TSH analogs on the induction of 18 F-FDG uptake in FRTL-5 cells, when TR-1401 displayed a 48-fold increase and TR1402 showed a 4.4-fold increase in potency compared with hTSH (Fig. 3, Table 1). TR1401 and TR1402 revealed similar potency in comparison with bTSH, which is known for its superagonistic potency toward TSH receptor-mediated effects. Using a defined number of FRTL-5 cell “batches” (see Statistical Analysis), the analysis of the maximal efficacy of the various TSH analogs for inducing 18 F-FDG uptake did not show any significant differences (Table 1).
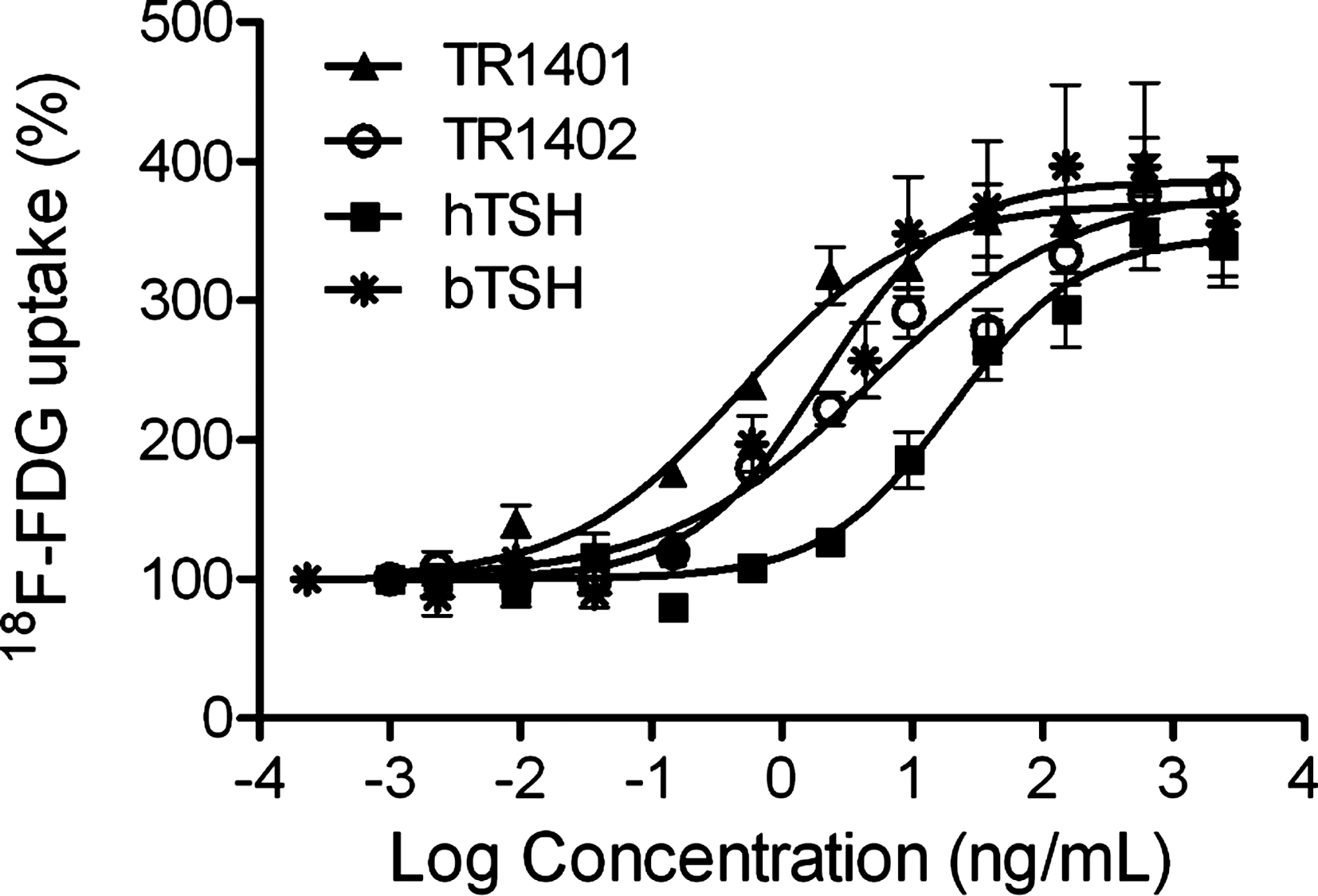
Uptake of 18F-FDG in stimulated FRTL-5 thyroid cells. Values are mean ± standard error (SEM) of 3–5 independent experiments performed in triplicate (n = 9–15). 18F-FDG, 18F-fluorodeoxyglucose.
Calculated values from data presented in Figure 3.
hTSH, human thyroid-stimulating hormone; bTSH, bovine TSH.
The specific PI3-kinase inhibitor LY294002 inhibited 18 F-FDG uptake by 63% ± 18% (n = 8) in TR1401-activated cells and by 36% ± 5% (n = 9) in TR1402-activated cells (Fig. 4). This influence was comparable to the inhibitory effect of LY294002 on stimulation by hTSH (56% ± 15%, n = 9) and confirmed our results on bTSH [58% ± 9% (31)].
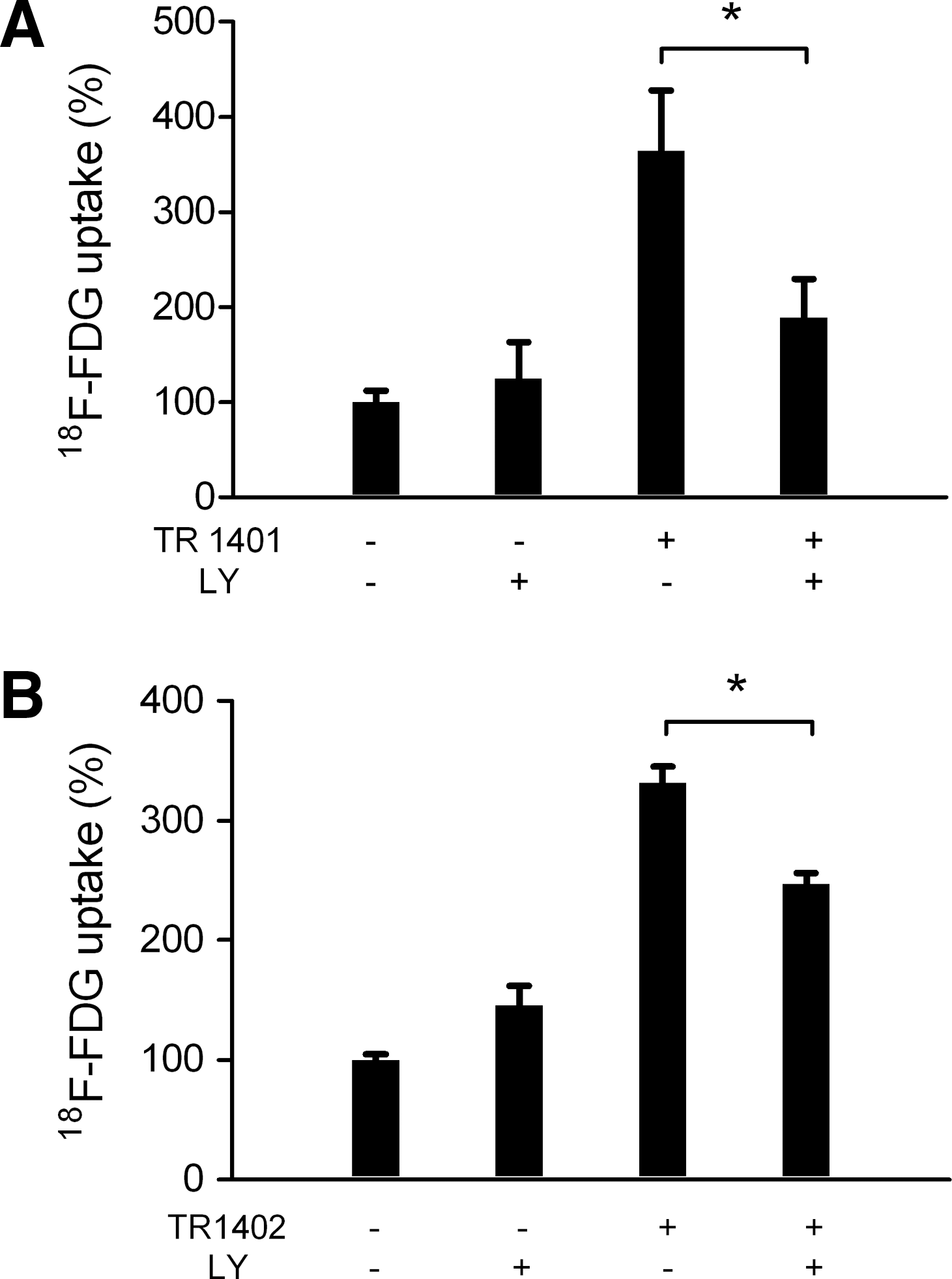
Inhibition of 18F-FDG uptake by the PI3K inhibitor LY294002 (10 μM, 30 minutes) before stimulation with TR1401
Concerning the activation of radioiodide transport in FRTL-5 cells, TR1401 and TR1402 displayed a 490-fold and 85-fold increase in potency in comparison with hTSH (Fig. 5, Table 2). Notably, the reason for the shallow slope of the TR1401 response curve for radioiodide uptake remained unknown. However, the EC50 values for TR1401 and TR1402 were not significantly different, due to the high standard deviation of the means (Table 2). Compared with hTSH, TR1401 did not increase the maximal efficacy (Emax) for the stimulation of radioiodide uptake. In contrast, TR1402 produced a 30% increase in maximal efficacy for the stimulation of radioiodide transport in FRTL-5 cells (Table 2), that was determined to be significant with a p-value of 0.05. The effect of TR1402 was comparable to that of bTSH, both in potency and in maximal efficacy (Table 2).

Uptake of 131I-iodide in stimulated FRTL-5 thyroid cells. Values are mean ± standard error (SEM) of 3–7 independent experiments performed in triplicate (n = 9–21).
Calculated values from data presented in Figure 5.
To verify the role of the PKA pathway known to mediate TSH-induced NIS expression and thus increased iodide transport, we used the selective PKA inhibitor H89 for studying its effect on iodide transport in TR1401, TR1402, bTSH, and hTSH-treated cells. As shown in Figure 6, the inhibition pattern of TR1401 and TR1402 was comparable (TR1401: 104.8% ± 2.2%, n = 6; TR1402: 95.7% ± 11.5%, n = 12), revealing almost complete inhibition of iodide transport by the PKA inhibitor. Similar results were obtained for inhibition of hTSH- or bTSH-induced iodide transport (hTSH: 91.5% ± 27.3%, n = 9; bTSH: 83.6% ± 24.2%, n = 9; data not shown in Fig. 6).
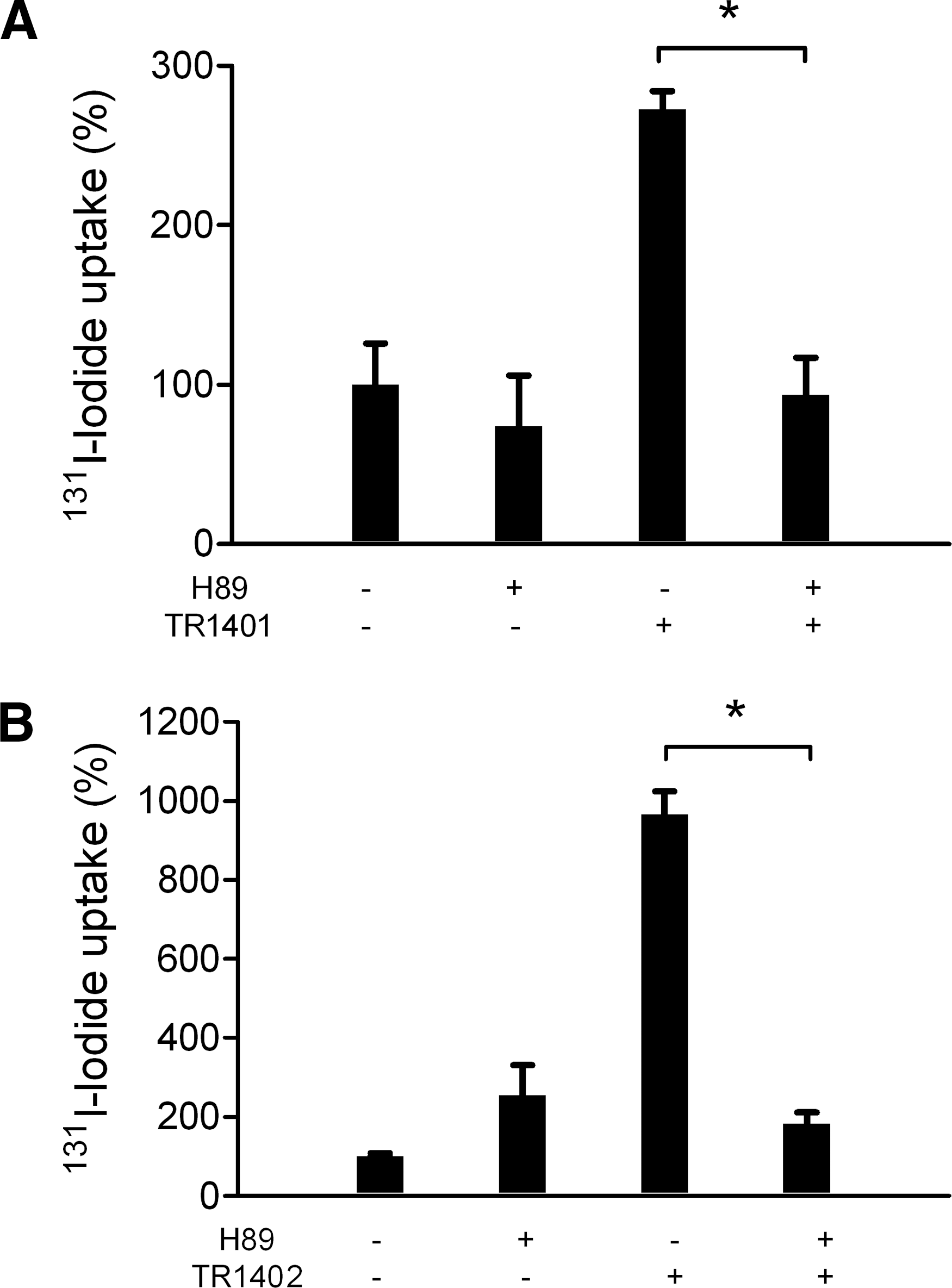
Inhibition of 131I-iodide uptake by the PKA inhibitor H89 in TR1401-stimulated
To determine the effect of TR1401 and TR1402 on the protein level for GLUT-1 and NIS expression in FRTL-5 cells, membrane fractions of TR1401- and TR1402-treated and untreated cells were studied by Western blot analysis (Fig. 7). We observed an increase in both NIS and GLUT-1 expression, reflecting the results of TR1401- and TR1402-induced radioiodide and 18 F-FDG uptake. Of note, the pronounced effect of TR1402 on the maximal efficacy of iodide transport was paralleled by a more than 25-fold increase in the membrane expression of NIS (Fig. 7).

Representative Western blot analysis of GLUT-1 expression and NIS expression in TR1401-treated
The biodistribution study using T3-suppressed CD-1 mice showed that radioiodide incorporation differed significantly between PBS-, TSH-, and TR1401-treated animals at all time points (Fig. 8), as determined by one-way ANOVA calculations. It is evident from the time–activity curves that the PBS-treated mice exhibited very low radioiodide uptake in the initial uptake phase (0–12 hours; Fig. 8) and throughout the 48-hour uptake period (data not shown). The three different dose groups (3, 10, and 30 μg, each per animal; n = 4 for each time point p.i.) showed a time–activity curve that followed a one-phase exponential association that allowed the one-parameter curve fitting for the maximum uptake value (%ID/organ) by nonlinear regression (R 2 > 0.6). Importantly, differences in the entire curve were only observed between the 3 μg dose group of TR1401- and hTSH-treated animals as determined by the extra sum-of-squares F-test. Comparing the best-fit values for the maximum iodide uptake, it was evident that the 3 μg dose of TR1401 significantly increased the iodide uptake value by a factor of two when compared with hTSH (p < 0.05; Table 3). The total uptake ratios (TR1401/hTSH) were determined from the area under the curve (AUC) for the uptake period of 0–12 hours p.i. as a measure of absorbed radiation dose in the thyroid (Table 3). It is important to note that the low dose of 3 μg of TR1401 is superior to an equal dose of hTSH, since TR1401 generates twice the area under the curve for uptake of radioiodide than hTSH. Ten micrograms is the dose at which hTSH and TR1401 have equal efficacy, since the best-fit value of maximum uptake did not significantly differ between TR1401 and hTSH. Additionally, the 30 μg dose produces an opposite result where hTSH provides almost twice the radioiodide uptake of TR1401. Thus, TR1401 showed the typical dose–response behavior of a highly potent activator, which becomes antagonistic at higher doses. These data show that TR1401 (3 μg/mouse) induced a superior radioiodide uptake and thus an increased absorbed radiation dose as compared with hTSH.
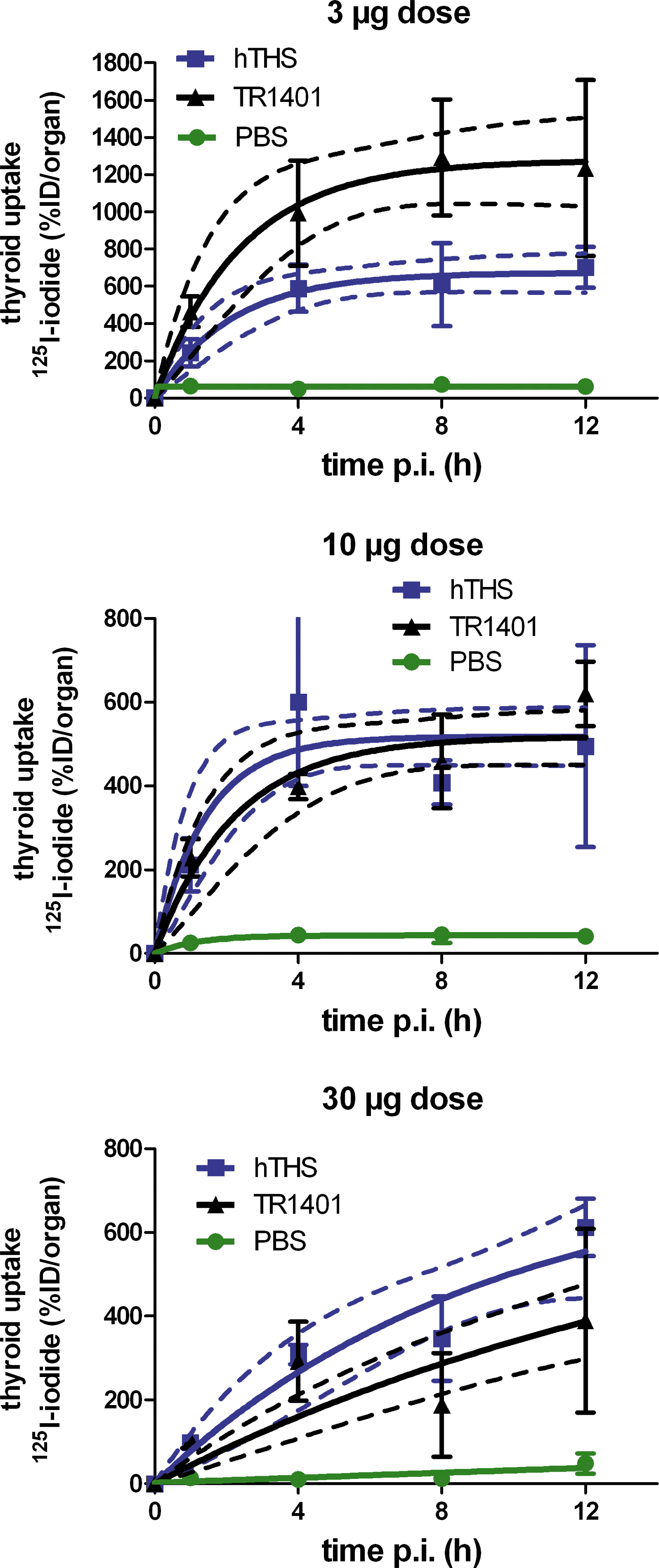
Ex vivo biodistribution time–activity curve of T3-suppressed CD-1 mice that were treated with TR1401, hTSH, or PBS alone in the 3, 10, and 30 μg dose groups. Data (%ID/organ) are presented as mean ± standard deviation (n = 4 for each time point and test compound, 0–12 hours p.i.). The curve fitting for one variable (max. uptake) by nonlinear regression using a one-phase exponential association kinetic was performed using GraphPad Prism software (GraphPad Software, San Diego, CA). The dashed lines represent the 95% confidence intervals for the best-fit value of maximum uptake.
The curve fitting for one variable (max. uptake) by nonlinear regression using a one-phase exponential association kinetic was performed using GraphPad Prism (vers. 5.00; GraphPad Software, San Diego, CA).
The unpaired t-test to compare the best-fit value of maximal uptake was used for the time–uptake curves of TR1401- and hTSH-treated animal groups (0–12 hours p.i., n = 4 for each time point p.i.). p-Values of <0.05 were considered to be significant.
AUC, area under the curve; ND, not determined.
In addition, PET/CT imaging studies using single animals also revealed the highest 124I-iodide uptake in the thyroid for the low-dose TR1401-treated mouse (3 μg/animal; 260%ID/g) in comparison with the corresponding hTSH treated mouse (3 μg/animal; 30%ID/g) and the 10 μg-treated animals (hTSH: 76%ID/g; TR1401: 52%ID/g) (Fig. 9). It is important to note that the iodide uptake values from PET imaging data for each drug at each dose reflects the measurements from single animals, which may fall within the error bars of averaged data collected from multiple animals. However, the PET data gathered using individual mice treated with T3 followed by 3 μg TSH/TR1401 or 10 μg TSH/TR1401 and the PBS control confirmed the general trend of the ex vivo biodistribution studies, indicating the highest radioiodide accumulation in the thyroid at maximally potent low doses of TR1401, when compared with the natural hormone hTSH.
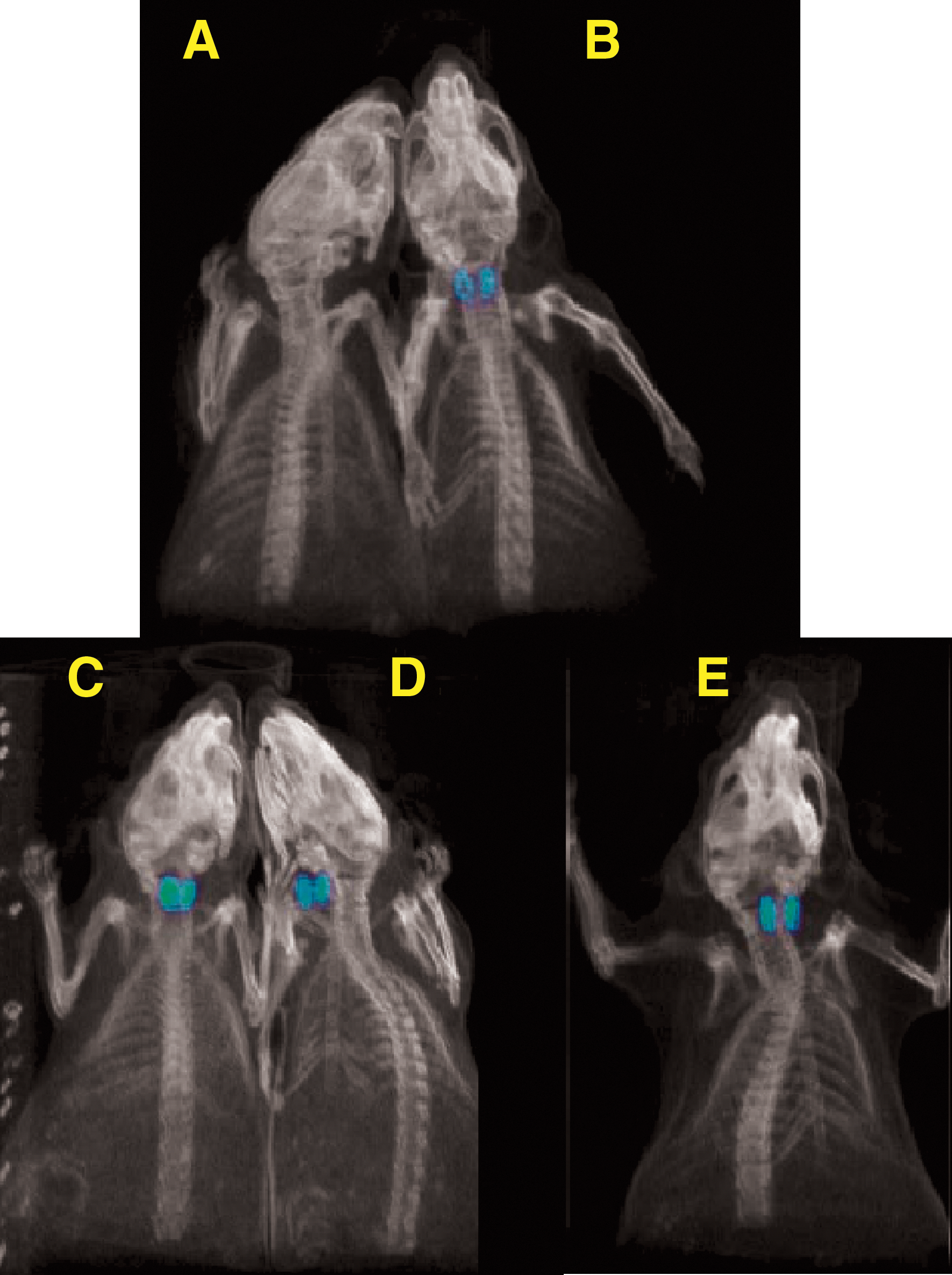
Positron emission tomography/computed tomography of CD-1 mice treated with PBS
Discussion
It is well known that bTSH has a higher affinity to the human TSHR and a higher intrinsic activity than hTSH (18,33). Some investigators attributed these differences to variations in purity or carbohydrate structures as well as to other posttranslational modifications of pituitary TSH preparations. Contrary to those hypotheses and somewhat surprisingly, Szkudlinski et al. have instead identified individual amino acids in the α subunit of the hormone molecule that are responsible for the higher bioactivity of bTSH (16). This is, in particular, conferred by the positively charged residues in the α-L1 in bTSH. As a proof of principle, superagonistic human TSH analogs have been developed by site-directed lysine-scanning mutagenesis (16,20), demonstrating higher potency in stimulating intracellular cAMP levels in CHO cells stably expressing the human TSHR than hTSH (16,20,23). Recent evidence indicated that charge–charge interactions between the positively charged lysine residues in bTSH (K15, K17, K20, K24), and lysine residues in TR1401 (K13, K14, K16, K20), and the negatively charged amino acid of the hinge positions E297, E303, and D382 of the TSHR are critical for their high-affinity binding and signaling (17,23).
TR1401 has been generated by introducing four lysine residues into the α1-loop of hTSH. The synthesis of TR1402 has followed the same principle, using, however, arginine instead of lysine (Fig. 1). No data on the biological properties of TR1402 have as yet been reported.
Both TR1401 and TR1402 displayed a significantly higher potency in stimulating uptake of radioiodine and 18 F-FDG in FRTL-5 compared with hTSH. This finding confirms the data on the superagonistic bioactivity of bTSH and TR1401 on the stimulating of cAMP levels (16,20,23), and extends our knowledge on these TSH analogs by focusing on the clinically relevant endpoints of radioiodide uptake and 18 F-FDG uptake. It is noteworthy that the enhanced bioactivity of highly purified TR1401 and TR1402 analogs is consistently shown in rodent and human TSH receptors in vitro and in vivo systems with various functional assay endpoints including deiodinase activity and thymidine incorporation in FRTL-5 cells [from ref. (16,20) and additional data not shown].
Interestingly, TR1402 showed a moderately higher efficacy in increasing radioiodide uptake than hTSH and TR1401. It is exciting to speculate on potential implications of observed discrepancy in signaling of two structurally very similar TR1401 and TR1402 analogs. On the basis of previous studies, it is anticipated that lysine to arginine substitution may result in higher TSH potency (20), but there have been no reports on difference in signaling pathways of such closely related human TSH analogs. Although additional studies are required to understand the relevance of such difference for future glycoprotein hormone design, it is likely that even slightly altered ligands can induce different hormone–receptor complex conformations leading to modified signaling, which is now known to be dependent not only on TSH receptor localized in the cell membrane, but also on TSH receptor signaling after internalization in the endocytic compartment (34). Thus, it is possible that observed differences in signaling between TR1401 and TR1402 analogs, including differences between cAMP stimulation and the relative potency to increase radioiodide or 18 F-FDG uptake, could be also a result of each analog preferential signaling in the plasma membrane and inside the thyroid cell. In addition, we cannot exclude that the 2-day exposure to TR1401 or TR1402 induced additional mechanisms that could reduce total accumulated radioiodide, such as, for example, affecting NIS protein expression. Although bTSH does not significantly modify pendrin gene expression in FRTL-5 cells (35), it is also possible that TR1401 could initiate such an effect by analog-specific modification of hormone–receptor complexes.
It is also intriguing to speculate on the clinical potential of the TSH superagonists reported herein. Although we observed major differences in potency between the superagonists and the currently available recombinant hTSH in vitro, there were no major differences in maximal efficacy. Theoretically, this in vitro difference in potency could be compensated by using standard recombinant TSH at much higher doses compared with TR1401 and TR1402 to obtain a similar maximal stimulatory effect. However, the highest dose of standard recombinant TSH employed in the phase 1 dose-ranging study showed toxicity of headaches and nausea and thus a true maximal efficacy dose of the currently available drug may not be achievable in patients (12). Moreover, it is well known that values describing potency and efficacy are not only dependent on the particular agonists under study, but also on receptor density and the efficiency of the intracellular response cascade of the cellular system in which these values have been measured (36). FRTL-5 is a model of well-differentiated thyroid tissue, but in thyroid cancer, various phenomena of dedifferentiation have been reported so that the behavior of this cell line may not be fully representative for human thyroid cancer tissue. Specifically, the maximal efficacy of any superagonist with increased potency is known to increase as receptor number decreases (36). Indeed dedifferentiated thyroid neoplasms have been reported to exhibit decreased expression of the TSH receptor as well as of the NIS (3,37,38). Further, their intracellular signal transduction cascades are less efficient than those of benign tissue since they usually harbor mutated signal transduction proteins acting as oncogenes. The relationship between the expression of a Ras oncogene and decreased TSH responsiveness of 18 F-FDG uptake in a cell line derived from a dedifferentiating follicular carcinoma of human origin has indeed recently been demonstrated (31).
In assessing the therapeutic potential of any TSH preparation or analog, one must also be aware of the major differences in potency and maximal efficacy assessed in vitro compared with in vivo. Ours and other studies have shown that pharmacokinetic factors such as delayed metabolic clearance or delayed absorption prolonging plasma half-life increase in vivo potency and efficacy much more than would be predicted from in vitro studies (39 –41). For example, in comparing bTSH prepared from a pituitary source to recombinant TR1401 and TR1402 prepared from CHO cells, these studies suggest that the potency and efficacy of the pituitary preparation versus the recombinant analogs would be overestimated in vitro compared with in vivo. The bTSH used in the current study was purified from bovine pituitaries that are known to produce carbohydrate chains terminated with sulfated N-acetylgalactosamine residues and very little sialylated oligosaccharide chains (39). In contrast, TR1401 and TR1402 are both recombinant products expressed in CHO cells producing highly sialylated and no sulfated oligosaccharides. Such carbohydrate compositions were previously associated with markedly attenuated TSH binding and activation in vitro but increased potency and efficacy in vivo (39 –41).
In the present work, we found by ex vivo biodistribution and in vivo PET imaging studies that TR1401, at low doses, generates twice the thyroid uptake of radioiodide than the same dose of hTSH. However, increasing the dose to 10 μg or 30 μg resulted in an opposite effect, which demonstrated the typical and well-known dose–response/negative-feedback behavior of a superagonist TSH analog that inhibits its induced effect at higher doses. Contrary to TR1401, hTSH clearly showed the near-linear effect of a typical low-affinity agonist in vivo as the injected dose increases. Due to the superagonist properties, TR1401 did not provide any higher radioiodide uptake beyond the 3 μg/mouse dose. As the maximal efficacy of any superagonist is known to increase as receptor density decreases, TR1401 could also be valuable when low TSH receptor densities, such as in dedifferentiated thyroid neoplasms, have to be addressed in vivo.
In addition, TR analogs have been shown in preliminary studies to cause delayed absorption after subcutaneous injection in mice and thus prolong plasma half-life even further when compared with equally sialylated recombinant wild-type TSH. Further in vivo studies in animal models of thyroid cancer are in progress and will be needed to confirm that these additional advantageous pharmacokinetic properties of TR analogs enhance their maximal efficacy for in vivo radioiodine uptake compared with wild-type recombinant TSH. On the basis of the evidence presented in this article, the clinical use of TR1401 and, particularly of TR1402, to increase radioiodine accumulation in de-DTC seems well founded.
Conclusions
TR1401 and TR1402 have significantly higher potency than hTSH to stimulate thyroidal iodine and fluorodeoxyglucose uptake in vitro. Moreover, ex vivo biodistribution and in vivo PET imaging studies indicated that at low doses, TR1401 induced an enhanced ability for the thyroid to concentrate iodide compared with the natural hormone hTSH. These properties as well as their lower immunogenicity compared with bTSH makes them interesting candidates for use in humans to enhance uptake of radioiodine and 18 F-FDG by metastases and recurrences of thyroid carcinoma.
Footnotes
Acknowledgments
The authors acknowledge Dr. Meng Zhang, Geping Wang, Shu-hui Huang, and Judy Klang at Trophogen, who contributed to the development, production, purification, and characterization of TR1401 and TR1402 analogs.
Disclosure Statement
V.F., B.D.W., and M.W.S. are current employees and V.W. is a former employee of Trophogen, Inc., who developed the TSH analogs used in the current study for commercial purposes and have a potential conflict of interest. However, Trophogen provided no funding for this study, which was performed independently by the other authors who have no conflict of interest.