
Abstract
Background:
Novel molecularly targeted drugs are undergoing preclinical and clinical testing to assess their efficacy against refractory thyroid carcinomas. The multikinase inhibitor Sunitinib has been shown to inhibit the kinase activity of the RET oncogene and reduce proliferation in differentiated thyroid cancer cells harboring the RET/PTC rearrangement. In this study, we evaluated its effects in human cell lines derived from differentiated (TPC-1) and anaplastic (8505C, CAL–62, and C643) thyroid cancers.
Methods:
The cells exposed to various concentrations of Sunitinib were examined for: (1) cell viability and presence of apoptosis, analyzed by cell counts, MTT assay, trypan blue exclusion assay, western blotting, and immunofluorescence; (2) expression of cyclin D1 and phosphorylated and nonphosphorylated extracellular signal-regulated kinase (ERK) and Akt proteins, analyzed by western blotting; and (3) transcription of genes encoding thyrocyte differentiation markers (thyroid-stimulating hormone receptor, sodium/iodide symporter, thyroglobulin, and thyroperoxidase) and proangiogenic factors (vascular endothelial growth factor A, platelet-derived growth factors A and B), measured by quantitative reverse transcriptase–polymerase chain reaction.
Results:
Exposure to nanomolar concentrations of Sunitinib significantly reduced cell viability in only TPC-1 cells, and this effect was paralleled by reduction of cyclin D1 levels. Western blotting revealed reduced phosphorylation of ERK and Akt after 3 and 6 hours of drug exposure. In contrast, the growth of 8505C, CAL-62, and C-643 cells was significantly reduced only by micromolar concentrations of Sunitinib, mainly due to induced necrotic rather than apoptotic death. In these cells, Sunitinib exerted a few significant effects on the transcription of angiogenic factors or thyrocyte differentiation markers.
Conclusions:
Sunitinib has little or no effect on the growth or differentiation of anaplastic thyroid cancer cells, thus suggesting that it is unlikely to be effective in the treatment of anaplastic thyroid cancer.
Introduction
Promising results have recently been obtained in some patients with combined therapies that include novel molecularly targeted agents (6 –8). Those directed against the protein tyrosine kinases (PTKs) are among the most promising. Thyrocyte growth regulation involves the activation of various PTKs, including growth factor receptors and elements of signal transduction pathways whose deregulation seems to be responsible for the cells' neoplastic transformation (9). Sunitinib, one of the several PTK inhibitors currently being tested in clinical trials, has produced encouraging results in patients whose carcinomas derived from thyroid follicular epithelial cells, including those with the more aggressive Hurthle-cell histotype (10 –12), but no information has been published on its efficacy in poorly differentiated and anaplastic thyroid carcinomas. Sunitinib is a multikinase inhibitor that acts mainly by antagonizing vascular endothelial growth factor receptors (VEGFRs) and platelet-derived growth factor receptor (PDGFR) kinases (13). However, it also suppresses the PTK-mediated proliferation and survival of tumor cells (14). As for thyroid carcinomas, Sunitinib exerts antiproliferative effects in thyroid carcinomas and promotes NIS gene expression in papillary thyroid cancer cell lines harboring the RET/PTC1 rearrangement (15).
In this preclinical study, we analyzed the effects of Sunitinib on growth and differentiation of 8505C, CAL-62, and C-643 cells, derived from human anaplastic thyroid cancer (16). As a control, we used TPC-1 cells, a widely used cellular model of papillary thyroid cancer with the RET/PTC1 rearrangement, which has already been confirmed as a target of this PTK inhibitor (17).
Materials and Methods
Cell cultures and analysis of cell viability and apoptosis
TPC-1 cells were provided by Prof. A. Fusco (University of Naples, Italy), and the 8505C cells were a gift from Dr. C. Nucera (Harvard Medical School, Boston, MA). CAL-62 cells were purchased from DSMZ GmbH (Braunschweig, Germany), and C-643 cells were provided by Nils Erik Heldin (University of Uppsala, Sweden). The thyroid origin of all four cell lines has recently been proved by genotype analysis (18). The cells were grown as previously described (16,19 –21) and treated with Sunitinib (from Pfizer Central Research, Groton, CT) diluted in dimethyl sulfoxide (DMSO). Controls were exposed to DMSO alone at the same concentration present in cultures exposed to 10 μM of Sunitinib.
For growth rate analysis, cells were seeded into 12-well plates (6×103 cells/well). The next day, the growth medium was replaced with fresh medium (untreated cultures) or medium containing Sunitinib at concentrations ranging from 0.001 to 10 μM. Cells were harvested after 24, 48, and 72 hours of incubation and counted in a Neubauer chamber. Proliferation was also evaluated with the 3-(4,5,-dimethylthiazole-2yl)-2,5-diphenyltetrazolium bromide (MTT) method (22). For this assay, 3×103 cells were seeded into 96-well plates in 100 μL of medium. Twenty-four hours later, the medium was changed, Sunitinib or DMSO (control) was added, and the plates were incubated for 24, 48, and 72 hours. The solubilized formazan product was quantified with a microplate spectrophotometer (Multiskan MS 6.0; Labsystems) at a wavelength of 540 nm and a reference wavelength of 690 nm. Cell mortality was evaluated by trypan blue exclusion, as previously described (22).
Apoptosis was detected by assessing the cleavage of caspase 3 by western blot analysis. In addition, analysis of nuclei morphology was performed by fluorescence microscopy after staining of the cells with 1 μg/mL Hoechst 33258 (Bio-Rad Laboratories s.r.l., Milan, Italy). Briefly, cells were seeded on glass cover slips and incubated with Sunitinib 5–10 μM for 72 hours or with Doxorubicin 1 μM for 24 hours. After two washes with PBS, cells were fixed with 4% paraformaldehyde for 20 minutes at 37 °C, washed with PBS, and stained with 1 μg/mL Hoechst 33258 for 20 minutes at 37 °C in the darkness. After two PBS washes, the cells were mounted on slides with Slow-Fade reagent (Molecular Probes, Leiden, The Netherlands) and observed with Nikon Diaphot fluorescence microscopy at an excitation wavelength of 365 nm (22).
Protein extraction and western blot analysis
Total proteins were extracted from the thyroid cells as previously described (23). Briefly, confluent cells from a single Petri dish were collected; homogenized in 150 μL of buffer containing NaCl 0.15 mM, MgCl2 1.5 mM, HEPES 50 mM, EGTA 5 mM, glycerol 1%, triton 1% (all from Sigma-Aldrich S.r.l. Milan, Italy), and complete protease inhibitor cocktail (Roche Diagnostics, Milan, Italy); and centrifuged at 135g (4°C for 10 minutes). The supernatant containing the whole-cell lysate was then centrifuged at 1352g (4°C for 15 minutes) and spectrophotometrically quantified with the Bradford method. Thirty micrograms of protein were loaded onto a 10%–12% SDS-polyacrylamide gel and subjected to electrophoresis at a constant voltage (120 V). Proteins were transferred to Hybond ECL-PVDF membranes (G.E. Healthcare, Milan, Italy) with the Mini Trans Blot system (Bio-Rad Laboratories S.r.l.; 2 hours at 225 mA). Membranes were blocked with Tween–tris buffered saline (TTBS)/milk (TBS, 1% v/v Tween 20, and 5% w/v nonfat dry milk) for 1 hour at room temperature and incubated with each of the following antibodies: affinity-purified polyclonal anticyclin D1 antibody diluted 1:500 (Santa Cruz Biotech, Santa Cruz, CA), polyclonal anti-Akt and anti-p-Akt antibodies diluted 1:200 (Santa Cruz Biotech.), polyclonal anti-ERK1,2 and anti-p-ERK1,2 antibodies diluted 1:1000 (Cell Signaling Technology, Danvers, MA), and monoclonal anticaspase 3 antibody (Cell Signaling Technology) diluted 1:1000. For normalization purposes, immunoblots were incubated overnight at 4°C with mouse monoclonal anti-human β-actin antibody (Sigma-Aldrich S.r.l.) diluted 1:5000 in TTBS/milk. After one 15-minute and two 5-minute washes in TTBS, the membrane was incubated with horseradish peroxidase-conjugated anti-rabbit (Sigma-Aldrich S.r.l.) or anti-mouse antibody (Transduction Laboratories, Lexington, KY) diluted 1:5000 and 1:10,000 in TTBS/milk, respectively. After one 15-minute and two 5-minute washes in TTBS, the protein was visualized with an enhanced chemiluminescence western blot detection system (ECL Plus; G.E. Healthcare).
RNA isolation, reverse transcription, and quantitative real-time polymerase chain reaction analysis
Levels of thyroid-stimulating hormone receptor (TSHR), NIS, Thyroglobulin (Tg), Thyroperoxidase (TPO), VEGF-A, PDGF-A, and PDGF-B mRNA were determined with real-time quantitative reverse transcriptase–polymerase chain reaction (RT-PCR), as previously described with minor modifications (24). Briefly, the Trizol method (Invitrogen, Carlsbad, CA) was used according to the manufacturer's instructions to extract total RNA from cells cultured with or without Sunitinib. One microgram of RNA was then reverse transcribed in a 20 μL reaction volume by using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. The complementary DNAs (cDNAs) were then diluted 1:5 in nuclease-free H2O (GIBCO, Milan, Italy) and amplified in an Applied Biosystems 7900HT Fast Real-Time PCR Sequence Detection System (Applied Biosystems) by using fast quantitative PCR thermal cycler parameters. In accordance with the manufacturer's instructions, each tube (reaction mixture volume: 20 μL) contained 2.0 μL of cDNA, 10.0 μL TaqMan Fast Universal PCR master mix (Applied Biosystems), and 1.0 μL of a predeveloped primer/probe mixture for each of the genes evaluated (FAM dye-labelled probes, Assay-on-Demand Gene Expression Products, and Applied Biosystems). Endogenous control (β-actin) was purchased from Applied Biosystems as predeveloped TaqMan Assay Reagents (VIC dye-labeled). All amplification reactions were performed in triplicate; the threshold cycle (C t) was obtained with Applied Biosystems software (SDS version 2.3) and averaged. Results were determined by the 2−ΔΔCt method (24) and expressed as relative expression normalized to a sample of control cells.
Statistical analysis
The results of cell growth experiments are expressed as means±SD. Differences between data collected at various experimental time points were evaluated with one-way ANOVA followed by the Turkey–Kramer multiple comparisons test performed with GrafPAD Software for Science (San Diego, CA). Quantitative RT-PCR results are expressed as means±SD, and the significance of differences was assessed with the t-test. p-values<0.05 were regarded as statistically significant. Data analysis was performed by using StatView 5.0.1 software (SAS Institute Inc., Cary, NC).
Results
Effects of Sunitinib on the growth of human thyroid cancer cells
As shown in Figure 1, exposure to nanomolar concentrations of Sunitinib significantly inhibited the growth of TPC-1 cells in a dose- and time-dependent manner. The maximal cell-count reduction was observed after 72 hours of exposure (Fig. 1A). In contrast, the proliferation of 8505C cells was significantly inhibited only after 72 hours of exposure to micromolar concentrations of Sunitinib. Even lower effects were detected in C-643 and CAL-62 cells with the maximal inhibition of cell growth only after 72 hours of exposure at 10 μM (Fig. 1A). Similar results were observed in all cell lines when the compound's antiproliferative effects were assessed with the MTT assay (data not shown). Thus, an EC50 of approximately 0.07 μM was detectable in TPC-1 cells, whereas for the anaplastic cell lines, it was in micromolar ranges (Fig. 1A). The effect was not paralleled with increased cell death. In fact, only higher Sunitinib doses (≥5 μM) were able to exert a significant cytotoxic effect, as documented by trypan blue exclusion assay (Fig. 1B). Both evaluation of caspase-3 cleavage (Fig. 1C) and cell nuclei staining with Hoechst 33258 documented absence of apoptotic death in all cell lines. Exposure to 0.1 μM for 72 hours reduced cyclin D1 protein expression levels in TPC-1 but not in 8505C cells (Fig. 1D).
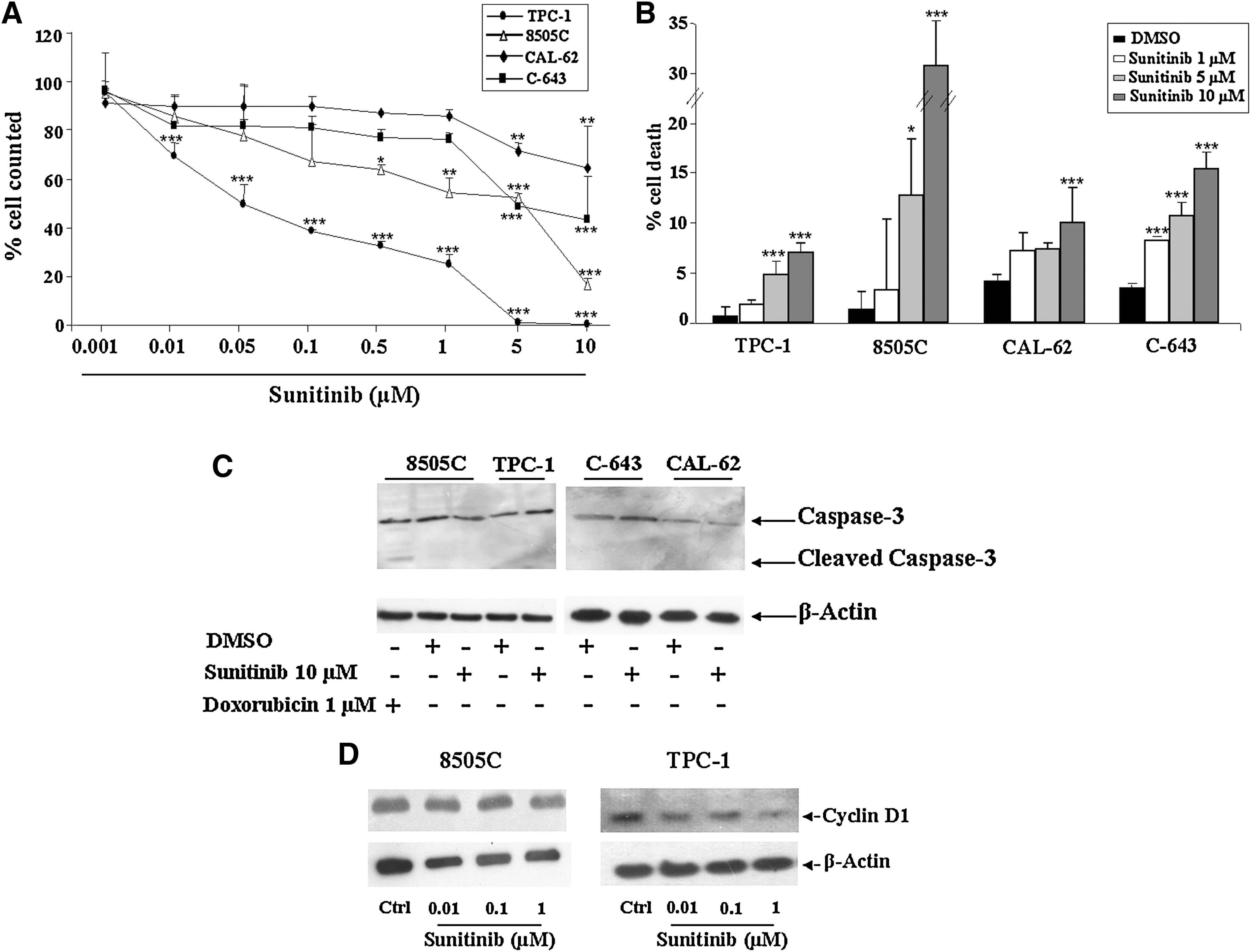
Effects of Sunitinib on proliferation and apoptosis on TPC-1, 8505C, CAL-62, and C-643 cells. Cells were treated for 72 hours with different concentrations of Sunitinib.
Effects of Sunitinib on growth-regulatory signal transduction pathways
Our next objective was to identify signal transduction pathways involved in Sunitinib's growth-inhibiting effects. To this end, we examined mitogen-activated protein (MAP) kinase and Akt-dependent signaling in the thyroid cancer cells. As shown in Figure 2, time-course experiments revealed clearly reduced phosphorylation of both Akt and extracellular signal-regulated kinase (ERK) in TPC-1 cells after 3 and 6 hours of exposure to Sunitinib (at doses ranging from 0.1 to 1 μM). At the 24–48 hours assessment, these effects were no longer seen. In contrast, phosphorylation of Akt and ERK in 8505C cells was unaffected by Sunitinib treatment at all the time points examined. In CAL-62 cells, an increase of phosphorylated ERK at 3–6–24 hours, a light reduction at 48 hours, and an increased expression of phosphorylated Akt at 3–6 hours, which then decreased, were observed. In C-643 cells, an increase at 3–6 hours with a following reduction at 24 and 48 hours of phosphorylated ERK was observed after exposure to Sunitinib, whereas there were no differences for phosphorylated Akt levels.
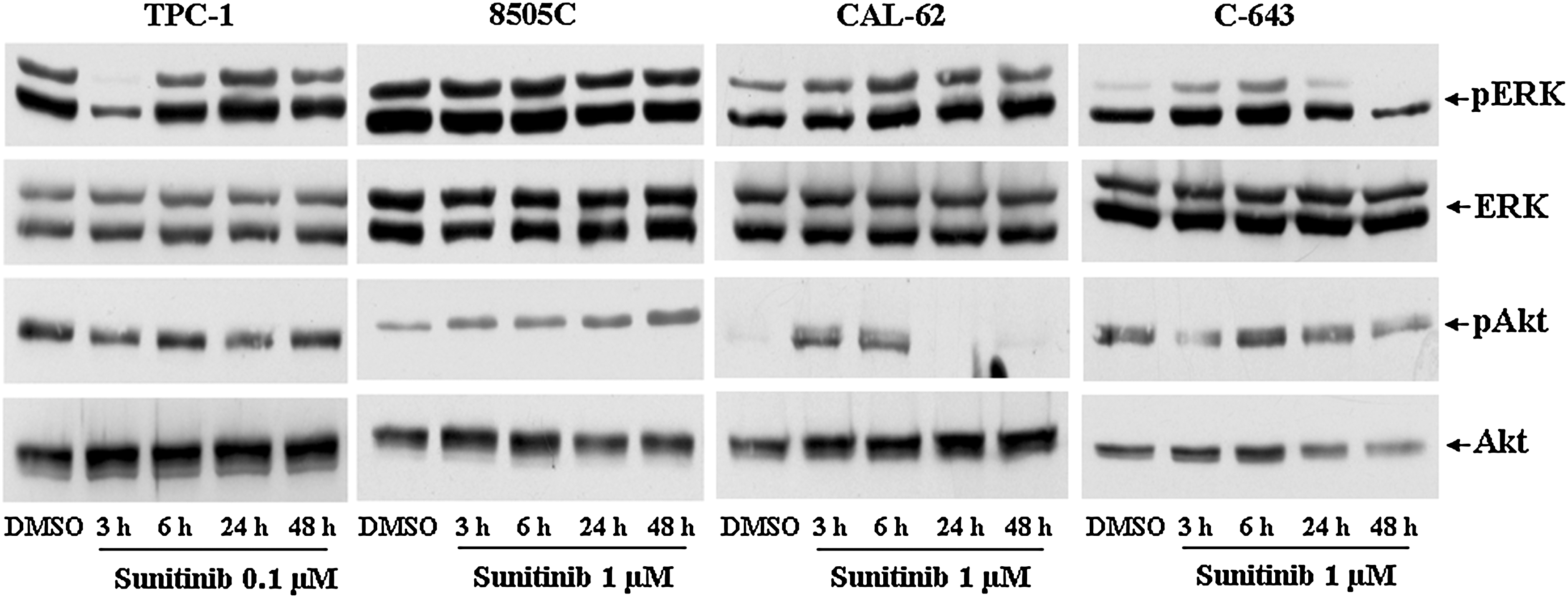
Effects of Sunitinib on extracellular signal-regulated kinase (ERK) and Akt phosphorylation in TPC-1, 8505C, CAL-62, and C-643 cells. Immunoblot analysis of ERK, p-ERK, Akt, and p-Akt expression in TPC-1, 8505C, CAL-62, and C-643 cells treated for 3, 6, 24, and 48 hours with Sunitinib. Experiments were performed as described in Materials and Methods section. Each result shown is representative of three different experiments.
Effects of Sunitinib on thyrocyte differentiation markers and angiogenic factors
We also examined the effects of Sunitinib on NIS, TPO, Tg, and TSHR gene expression in anaplastic thyroid cancer cells. Under basal conditions, these cells are characterized by undetectable expression of all these thyrocyte differentiation markers, except for Tg gene expression, just barely detectable. When cells were exposed for various periods to Sunitinib (at nanomolar and micromolar concentrations), the expression of the differentiation markers were below the detection threshold of our real-time PCR assays, with no signals at all after 35 cycles, except for Tg levels, which showed a threefold increase in CAL-62 and C-643 cells (Fig. 3 and data not shown).
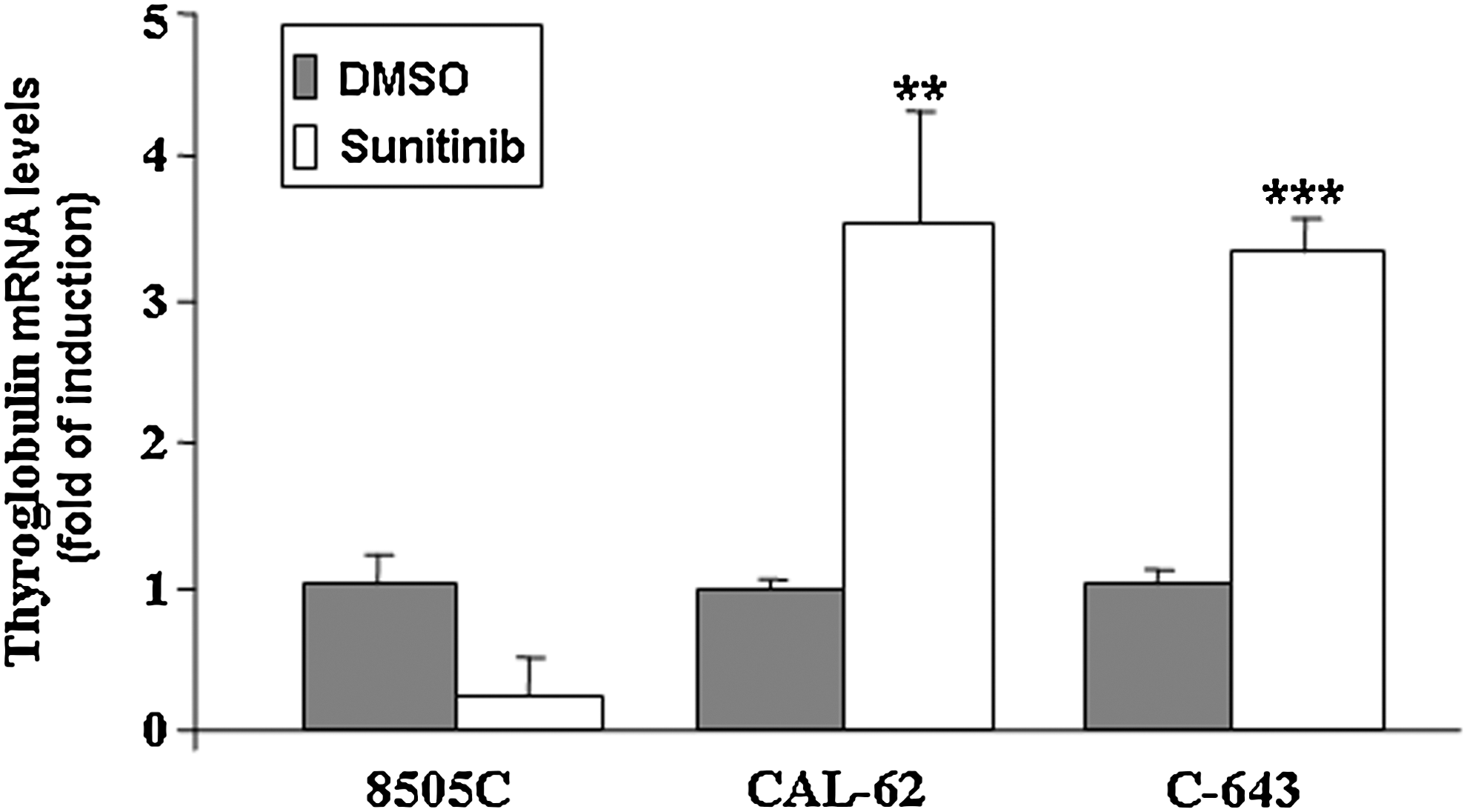
Effects of Sunitinib on the mRNA levels of Thyroglobulin in 8505C, CAL-62, and C-643 cells. Thyroglobulin mRNA levels were measured in 8505C, CAL-62, and C-643 cells treated for 72 hours with Sunitinib 1 μM. Total RNA was extracted as described in Materials and Methods section. β-actin was amplified as an endogenous control. Results are expressed as the mean±SD of values obtained from at least three different experiments. The t-test was used for statistical analysis. **p<0.01; ***p<0.001.
Finally, Figure 4 shows Sunitinib's effects on the transcription of genes encoding the proangiogenic factors VEGF-A, PDGF-A, and PDGF-B in anaplastic thyroid cancer cells exposed to 1 μM of Sunitinib for 72 hours. Any effect on transcript levels for these genes did not result in a statistically significant difference after Sunitinib treatment, except for an increase of all the proangiogenic factor mRNA observed in CAL-62 cells and of PDGF-B in C-643 cells (Fig. 4).
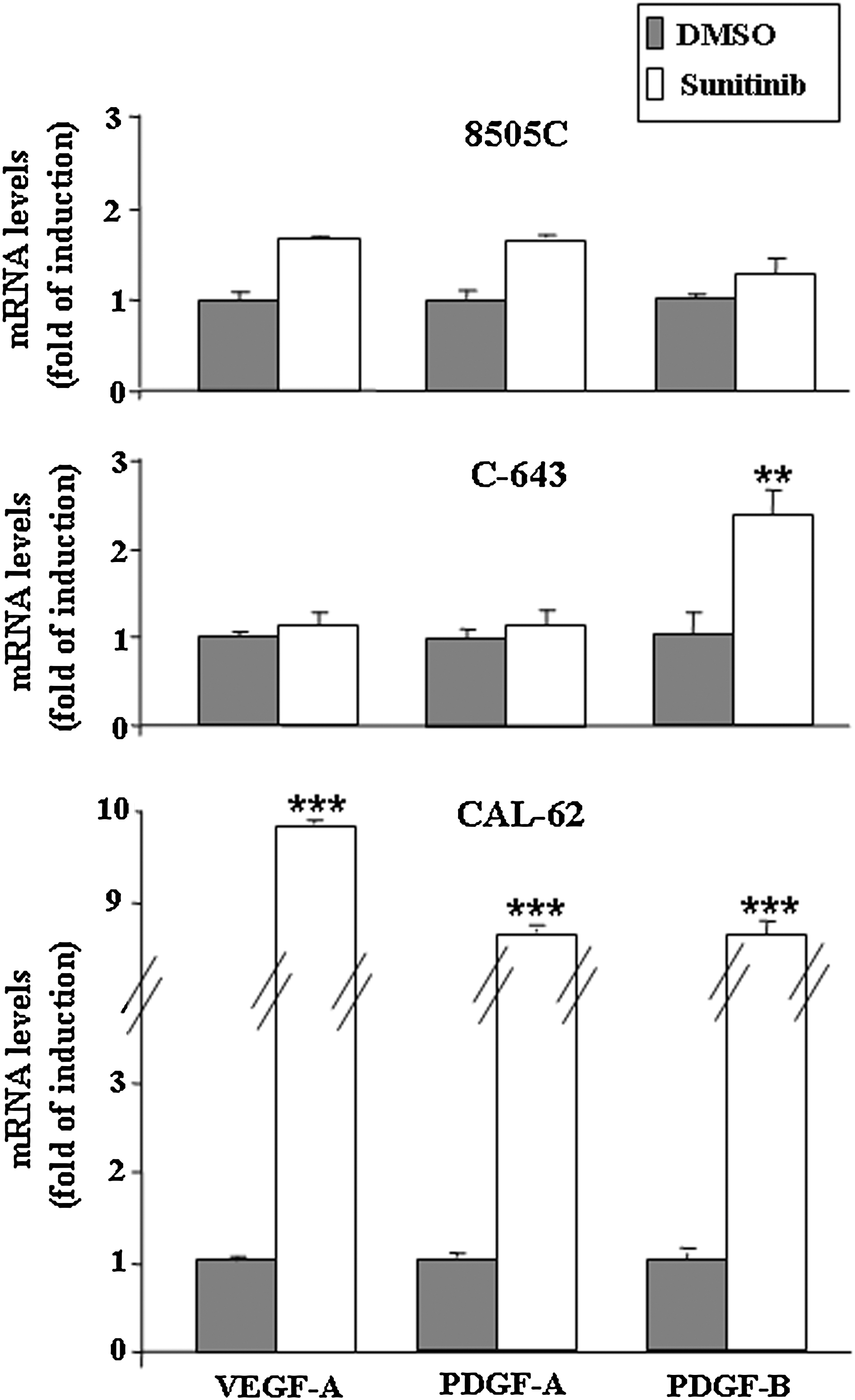
Effects of Sunitinib on the transcription of genes encoding proangiogenic factors in 8505C, CAL-62, and C-643 cells. VEGF-A, PDGF-A, and PDGF-B mRNA levels were measured in 8505C, CAL-62, and C-643 cells treated for 72 hours with Sunitinib 1 μM. Total RNA extraction and quantitative RT-PCR were performed as described in Materials and Methods section. Results are expressed as the mean±SD of values obtained from at least three different experiments. The t-test was used for statistical analysis. **p<0.01; ***p<0.001. VEFG, vascular endothelial growth factor; PDGF, platelet-derived growth factor; RT-PCR, reverse transcriptase–polymerase chain reaction.
Discussion
Novel drugs that suppress oncogene-activated PTK activity in cancer cells have produced very encouraging results in the treatment of solid tumors, including thyroid malignancies (5 –7,25). Particular interest has been focused on the multikinase inhibitor Sunitinib, whose performance in preclinical studies included the reduction of RET kinase activity and inhibition of proliferation in both normal (26) and neoplastic (15,17) thyroid cells. Data generated by clinical trials also support the compound's therapeutic potential in thyroid cancer. In two independent phase II studies, objective response rates ranged from 13% to 31% (12,10), and in a case recently reported by Kaldrymides et al., the drug also seems to have produced partial remission of metastatic papillary thyroid carcinoma (27). However, all these preclinical and clinical data alike refer to Sunitinib's effects on well-differentiated thyroid carcinoma histotypes. To date, its efficacy has never been specifically investigated in the treatment of poorly differentiated or anaplastic thyroid carcinomas.
For this reason, we explored the effects of Sunitinib in three cell lines derived from human anaplastic thyroid cancer, and compared them with those observed in TPC-1 cells, a thyroid cell line harboring the RET/PTC1 rearrangement. In the latter cells, nanomolar concentrations of the drug significantly reduced cell growth, which is fully consistent with the recently published data of Fenton et al. (15), whereas in all the anaplastic thyroid cells, antiproliferative effects were observed only with micromolar concentrations of Sunitinib, which also proved to be highly cytotoxic, as reflected by high cell death rates, mainly due to pro-necrotic rather than pro-apoptotic effect. In accordance with these findings, the drug had no appreciable effects on phosphorylation of ERK and Akt in 8505C cells, which reflect the activities of the two signaling pathways that are the main regulators of thyroid cell proliferation. Similarly, no effect, or eventually a marginal paradoxycal activation, on the MAPK and the PI3K-Akt pathways could be observed in CAL-62 and C-643 cells. Fluctuating levels of phosphorylated ERK and Akt protein, observed in our study, have been already reported when thyroid cancer cells were treated with protein-kinase inhibitors, probably due to the concomitant associated down-regulation of the phosphatases MKP-3 and DUSP5 (28). Altogether, these observations should be related to the peculiar mutations driving the neoplastic phenotype of 8505C cells and CAL-62 and C-643 ones, respectively, BRAF or RAS mutations, which do not represent known targets for Sunitinib.
Fenton et al. have also reported some degree of redifferentiation in papillary thyroid cancer cell lines exposed to very low concentrations of Sunitinib (15). An effect of this type could be very important for refractory thyroid malignancies, provided it included restoration of the iodide-concentrating capacity, as this would significantly increase the treatment options for these tumors (3). We, therefore, assessed the redifferentiating potential of Sunitinib in our cell model of anaplastic thyroid cancer. Unfortunately, the drug had no effect on the expression of NIS or the genes encoding other thyrocyte differentiation markers, except for Tg in two cell lines. This finding suggests that the drug's redifferentiating activity may be related to its inhibition of RET kinase, as documented by Kim et al. (17), and that it is exerted when this oncogene is the driver of tumorigenesis. Although all the anaplastic thyroid cells used in this study exhibit a variety of genetic and epigenetic alterations, RET activation has never been detected in these cell lines (18), and the same is true for human anaplastic thyroid cancers in general (5).
The anticancer effects exerted by Sunitinib in vivo are known to depend mainly on a reduction of tumor neo-vascularization secondary to the inhibition of proangiogenic factor release and activity (13,14). Reduced thyroid tissue vascularity has been recently reported in a patient receiving Sunitinib for the treatment of metastatic renal cancer (29), and increased VEGF expression has been documented in tumor tissues (30,31) and serum (32) from patients with advanced papillary thyroid cancers. Higher than normal expression levels of this proangiogenic factor have also been found in anaplastic thyroid carcinomas (33), thus suggesting that the VEGF signaling pathway is a rational target in the treatment of these tumors as well. However, in 8505C cells that harbor the BRAF V600E mutation (18), an alteration associated with high expression levels of VEGF-A levels in at least one study (34), as well as in C-643 cells, Sunitinib had no effect on the transcription of VEGF-A or PDGF-A and PDGF-B genes. A paradoxical increase of all the proangiogenic factors was observed in CAL-62 cells. However, according to our data, we cannot exclude the possibility that it does affect the activity of receptors for these proangiogenic ligands, which are mainly expressed in endothelial cells of the stroma adjacent to the tumor.
In conclusion, our data confirm that the PTKs inhibited by Sunitinib are valid targets in the treatment of differentiated thyroid tumors bearing the RET rearrangement. However, the drug seems to have little or no effect on proliferation, differentiation, and proangiogenic gene expression in anaplastic thyroid cancer cells, which suggests that it will be of limited value for treatment of this type of thyroid malignancy.
Footnotes
Acknowledgments
This work was supported by the Fondazione Umberto Di Mario ONLUS and by grants from the Fondazione Cassa di Risparmio di Perugia. M.D. and V.M. are fellows of the Ph.D. program of Pharmaceutical Sciences at the University of Catanzaro. The article was edited by Marian Everett Kent, who received payment from the sponsors for this contribution.
Disclosure Statement
No competing financial interests exist.