
Abstract
Background:
Thyroid-stimulating autoantibodies (TSAb) bind to the thyrotropin receptor (TSHR) extracellular domain, or ectodomain (ECD), comprising a leucine-rich repeat domain (LRD) linked by a hinge region to the transmembrane domain (TMD). The LRD (residues 22–260; signal peptide 1–21) contains two disulfide-bonded loops at its N-terminus. In the crystal structure of the isolated LRD complexed with human TSAb monoclonal antibody (mAb) M22, N-terminal disulfide loop 1 (residues 22–30) could not be determined because of crystal disorder. Nevertheless, present crystal structure data are interpreted to exclude a role for the LRD N-terminal disulfide loops in the TSAb epitope(s), contradicting prior functional evidence of a role for these loops in TSAb function.
Materials and Methods:
To re-examine this issue we studied two cell types expressing the TSHR with the extreme N-terminal loop 1 (residues 22–30) deleted: the TSHR ECD lacking the TMD and tethered to the plasma membrane by a glycosyl-phosphatidylinositol (GPI) anchor, and the TSH holoreceptor containing the TMD. Because TSAb including M22 “see” the holoreceptor poorly relative to the TSHR ECD-GPI, we used the latter to examine the effect of deleting residues 22–30 on M22 binding by flow cytometry and the holoreceptor to test the effect of this deletion on the functional response to M22.
Results:
Deletion of TSHR N-terminal loop 1 (residues 22–30) reduced the number of TSHR-ECD-GPI recognized by M22 relative to two TSHR mAb with epitopes far downstream of the LRD N-terminal loops. Relative to control mAb 2C11, M22 recognized only 60.4% of cell surface receptors (p = 0.02). In contrast to M22 binding to TSHR-ECD-GPI, in functional studies with the TSH holoreceptor, M22 stimulation of cAMP generation was unaltered by the loop 1 deletion.
Conclusions:
Our data support the concept that TSAb interact with the cysteine-rich N-terminus of the TSHR. Comparison of crystal structures of the same TSHR LRD in complex with TSAb M22 or blocking antibody K1-70 helps reconcile contradictory viewpoints. A difference between M22 interaction with the identical TSHR N-terminus expressed on the TSHR-ECD-GPI and holoreceptor suggests that crystallization of the TSHR LRD-M22 complex may not provide a complete understanding of the functional TSAb epitope(s) in Graves' disease.
Introduction
From their crystal structures, the TSHR N-terminus, immediately after the signal peptide (residues 1–21) containing a cluster of 4 cysteine residues at positions 24, 29, 31, and 41, forms two disulfide bonds (residues C24-C29 and C31-C41) (10,11). Remarkably, this order of cysteine linkage (1–2 and 3–4) forming two distinct loops (hereafter termed loop 1 and loop 2, respectively) is different to that in the closely homologous FSHR, in which the cluster of four cysteines are linked 1–3 and 2–4, forming a more tightly structured cysteine knot or “sushi domain” (9).
Studies from our laboratory over the past 20 years have suggested that the conformation of this N-terminal cysteine-rich region contributes to TSAb recognition and activation of the TSHR, as well as being highly immunogenic when mice are immunized with recombinant TSHR protein (12). For example, chimeric TSH-LH receptor 6-A1, in which TSHR amino acid residues SSPP in loop 1 were substituted with the corresponding rat LH receptor residues (HHRI), responded poorly to polyclonal TSAb in Graves' patients' sera (13) as well as to monoclonal human TSAb M22 (14). Further, differential recognition by polyclonal TSAb and a mouse monoclonal antibody (mAb) whose epitope included the TSHR cysteine-rich N-terminus revealed that this region contained two distinct conformational forms (15,16). On the other hand, deletion of the entire loop 1 (TSHR residues 22–30) had no effect on activation of the TSHR by polyclonal Graves' sera TSAb (17). The significance of these previous studies has recently been questioned. Thus, analysis of the crystal structure of M22 with the TSHR “showed that there were no M22 interactions involving the extreme N-terminal region of the TSHR LRD … not consistent with previous studies, which have concluded that the TSHR N-terminal region is part of a highly conformational epitope for thyroid-stimulating autoantibodies” (11).
In considering an explanation for these divergent conclusions, we hypothesized that, unlike the stable sushi domain in the FSHR, the homologous region of the TSHR with disulfide bonds between cysteines 1–2 and 3–4 would form a relatively unstable structure, allowing motion of the first loop (residues 22–29) relative to the second loop (residues 31–41) around a fulcrum of residue E30 (Fig. 1). Because of ambiguity in the TSHR N-terminal structural data (10), the discrepancy in the functional effect between loop 1 chimeric substitution and loop 1 deletion, and recent availability of monoclonal TSAb M22 (a much more powerful tool than polyclonal sera), in the present study we re-evaluated the effect of loop 1 deletion using TSAb M22 as a probe.
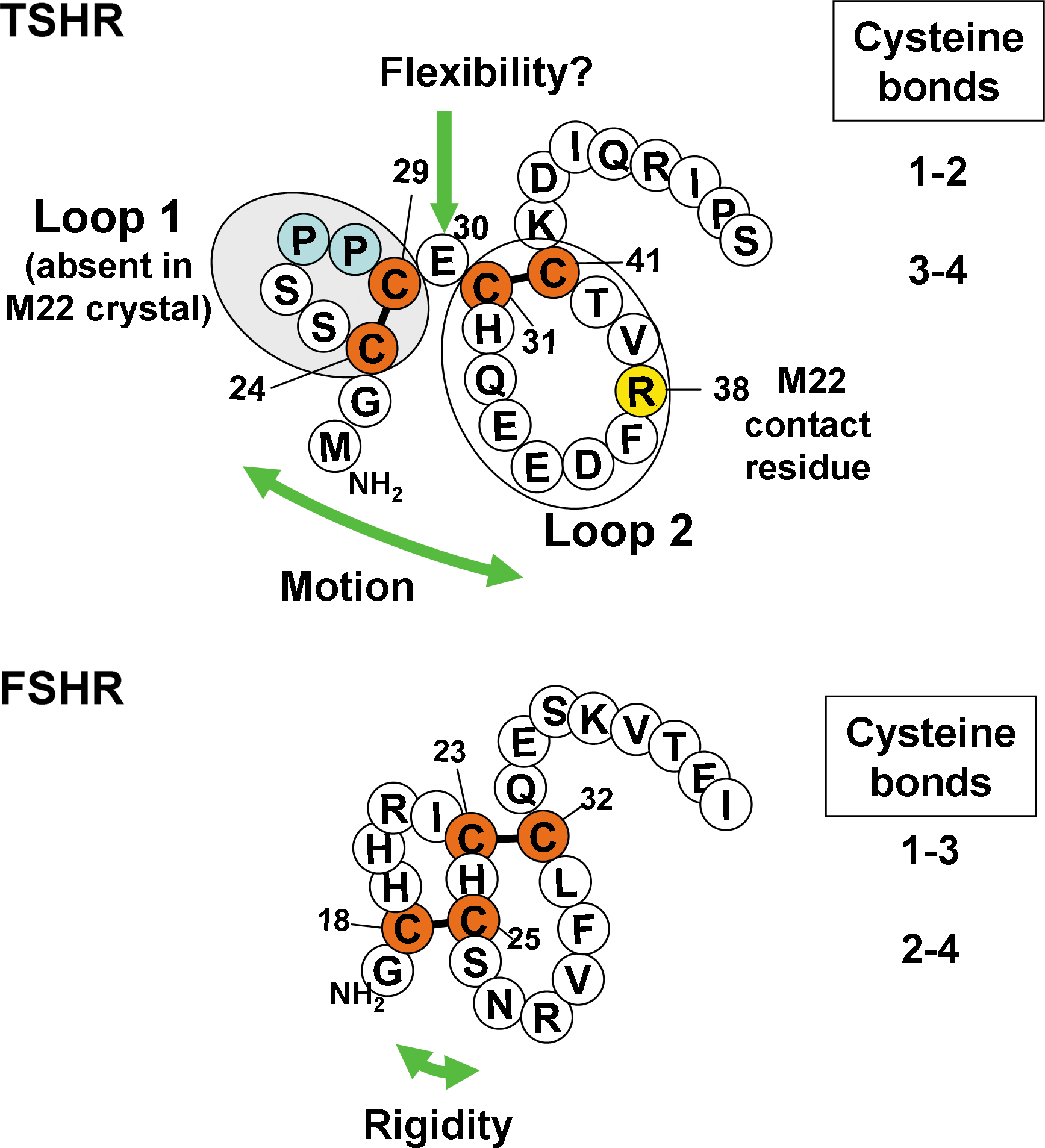
Hypothesis based on the secondary structures of the follicle-stimulating hormone (FSH) and thyrotropin (TSH) receptor N-termini. The glycoprotein hormone receptor N-termini have a cluster of four cysteine residues forming two disulfide bonds. The crystal structure of the leucine-rich repeat domain (LRD) of the FSH receptor (FSHR) in complex with FSH revealed that these disulfide bonds formed a sushi-like domain (1–3 and 2–4 cysteine linkages) (9). In contrast, in the TSH receptor (TSHR)-M22 crystal structure, a 3–4 (C31-C41) disulfide bond was observed (10). The structure upstream of C31 could not be determined because of molecular disorder; however, the 3–4 disulfide bonding indicates that cysteines 1 and 2 (C24-C29) are linked (10). Because disorder implies structural instability or movement, we hypothesized that the lack of a more rigid sushi-like domain in the TSHR permitted motion of disulfide linked loop 1 with flexibility centered on residue E30 bridging loop 1 and loop 2. M22 is known to contact residue R38 in TSHR loop 2 (10).
Materials and Methods
TSHR-expressing cells
We used stably transfected Chinese hamster ovary (CHO) cell lines expressing the following TSHR: (i) TSHR-ECD-GPI. Generation of this cell line was described previously (14). In brief, the TSHR ectodomain tethered by a glycosyl-phosphatidyl-inositol (GPI) anchor (plasmid kindly provided by Dr. A.P. Johnstone, London, United Kingdom) (18) was transferred into the expression vector pcDNA5/FRT (Invitrogen, Carlsbad, CA). (ii) TSHR Δ22–30-ECD-GPI. The SalI (blunted)-AflII fragment containing the TSH holoreceptor cDNA with codons 22–30 deleted was transferred into NheI (blunted)-AflII sites in TSHR-ECD-GPI. (iii) Wild-type TSH holoreceptor. Generation of these cells transfected with this cDNA in pcDNA5/FRT was described previously (14). (iv) TSHR Δ22–30 was generated by replacing the AflII-XbaI fragment in Δ22–30-ECD-GPI with the corresponding fragment of the wild-type TSH holoreceptor in the pcDNA5/FRT vector. Plasmids for newly generated cell lines were stably transfected using FuGENE HD (Roche, Indianapolis, IN) into Flp-In CHO cells (Invitrogen) according to the manufacturer's protocol except using reduced hygromycin B concentrations (50–100 μg/mL, increasing up to 200 μg/mL when tolerated). CHO cells were cultured in F12 supplemented with 10% fetal calf serum (FCS), 10 mM HEPES, 2 mM glutamine, and standard antibiotics.
Monoclonal TSHR antibodies
Human monoclonal TSAb M22 (19) was kindly provided by Dr. B. Rees-Smith (Cardiff, United Kingdom). Murine mAb 2C11 (20,21) was purchased from Serotec (Oxford, United Kingdom). Generation of murine mAb CS-17 has been reported previously (22).
Flow cytometry
TSHR-expressing CHO cells were harvested from 10-cm plates using 1 mM EDTA and 1 mM EGTA in phosphate-buffered saline (PBS). After washing twice with PBS containing 10 mM HEPES, pH 7.4, and 2% fetal bovine serum, the cells were incubated for 1 hour on ice in 100 μL of the same buffer with or without monoclonal TSHR antibodies at the concentrations indicated in individual experiments. After rinsing, the cells were incubated for 1 hour on ice with 100 μL FITC-conjugated goat anti-mouse IgG (1:100; Caltag, Burlingame, CA) or FITC-conjugated mouse anti-human IgG (1:5; BD Pharmingen, San Diego, CA). After rinsing, fluorescence was measured using a Beckman FACScan flow cytofluorimeter. Cells stained with propidium iodide (6 μg/mL final concentration) were excluded from analysis.
Cultured cell cAMP assays
TSHR-expressing CHO cells were transferred into 96-well plates (∼4 × 104 per well) 24 hours before assay. For bioassay, the culture medium was replaced with DMEM/F12 (1:1), 1 mM isobutyl-methylxanthine, 10 mM HEPES, 5% FCS, and the indicated concentrations of monoclonal TSAb or bTSH (Sigma-Aldrich, St. Louis, MO). After incubation with the cells for 2 hours at 37°C, the medium was aspirated and intracellular cAMP was extracted with 0.2 mL ethanol. The extracts were evaporated to dryness and resuspended in 0.2 mL of PBS, pH 7.5, and samples were assayed using the LANCE cAMP kit (PerkinElmer, Shelton, CT). The maximum cAMP responses attained were calculated using GraphPad Prism software (La Jolla, CA).
Results
Effect of deletion of TSHR N-terminal loop 1 on M22 recognition
Polyclonal human TSAb (23) and monoclonal TSAb M22 (14) preferentially recognize the TSHR ECD tethered to the plasma membrane by a GPI anchor in comparison to the TSHR holoreceptor in which the identical ECD is linked to the seven membrane-spanning helices. We therefore performed flow cytometry using CHO cells stably expressing the wild-type TSHR-ECD-GPI and the TSHR-ECD-GPI with N-terminal loop 1 (amino acids 22–30) deleted (Δ22–30-TSHR-ECD-GPI). As a control for TSAb M22 we included in parallel tubes another mAb, 2C11, with an epitope in the hinge region including amino acids 354–359, far downstream of the M22 epitope.
Cell surface fluorescence with increasing concentrations of 2C11 attained a plateau at 33 μg/mL with both the wild-type TSHR-ECD-GPI and the same receptor lacking N-terminal loop 1 (amino acids 22–30) (Fig. 2A). However, the deletion mutant expressed less well on the cell surface relative to the wild-type TSHR-ECD (Fig. 2A). With the wild-type TSHR-ECD-GPI, TSAb M22 attained a plateau at a far lower concentration (1 μg/mL) than 2C11 reflecting its very high affinity. However, relative to 2C11, M22 “saw” even fewer Δ22–30-TSHR-ECD-GPI than the wild-type TSHR-ECD (Fig. 2B). This surprising finding led us to repeat the study with another control TSHR mAb, CS-17, whose epitope is also distinct from that of M22, namely, on the convex surface of the LRD with a component in the hinge region. As with 2C11, in the same assay using parallel aliquots of cells, M22 recognized fewer molecules of the TSHR N-terminal deletion mutant on the cell surface relative to the wild-type receptor than did control mAb CS-17 (Fig. 3A, B).
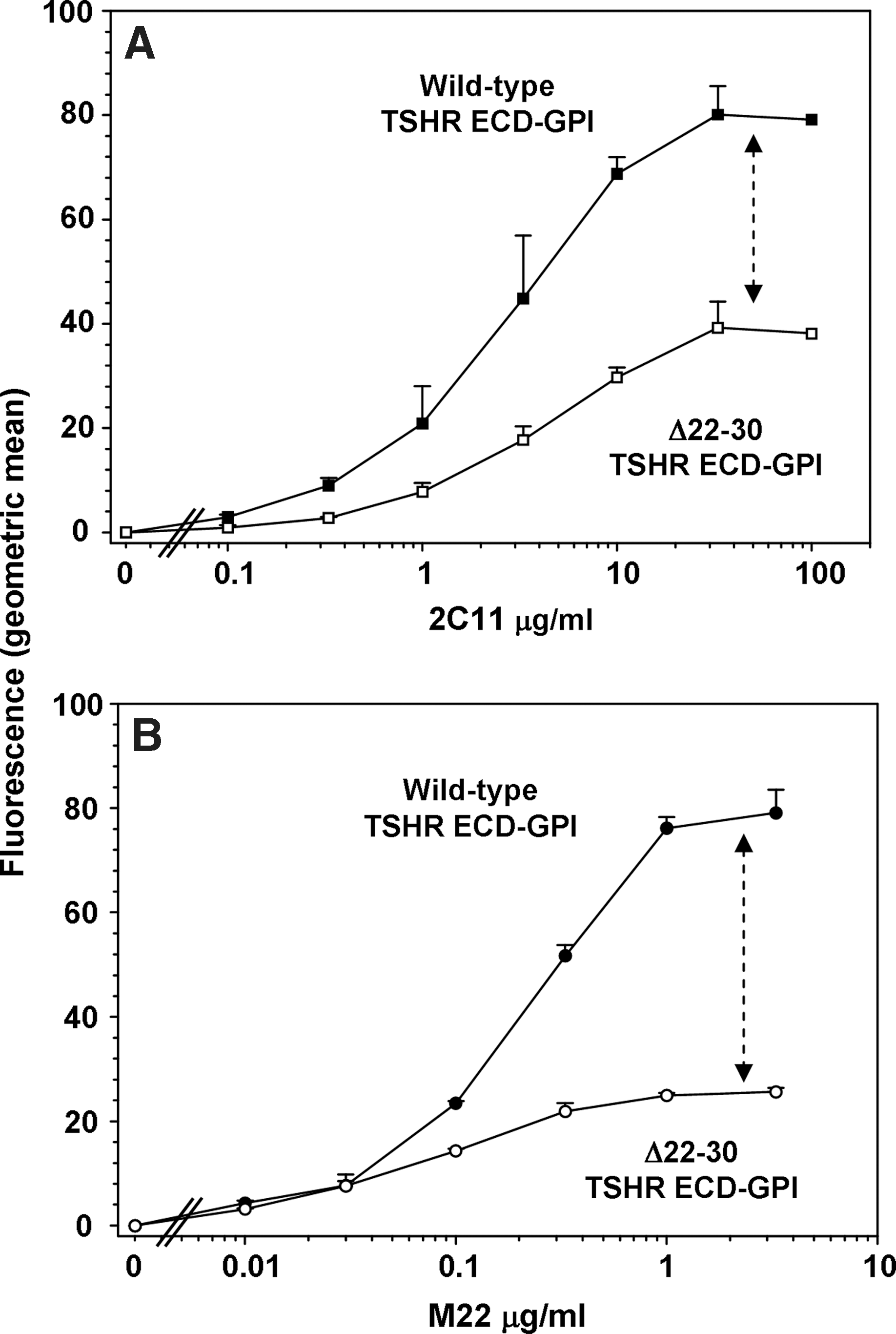
Deletion of TSHR N-terminal disulfide-linked loop 1 reduces the number of cell surface receptors detected by monoclonal human thyroid-stimulating autoantibody (TSAb) M22. Flow cytometry was performed using Chinese hamster ovary (CHO) cells stably expressing the wild-type TSHR ectodomain linked to the plasma membrane by a glycosyl-phosphatidyl-inositol anchor (TSHR-ECD-GPI) and cells expressing the same receptor with amino acid residues 22–30 deleted (Δ22–30 TSHR-ECD-GPI).
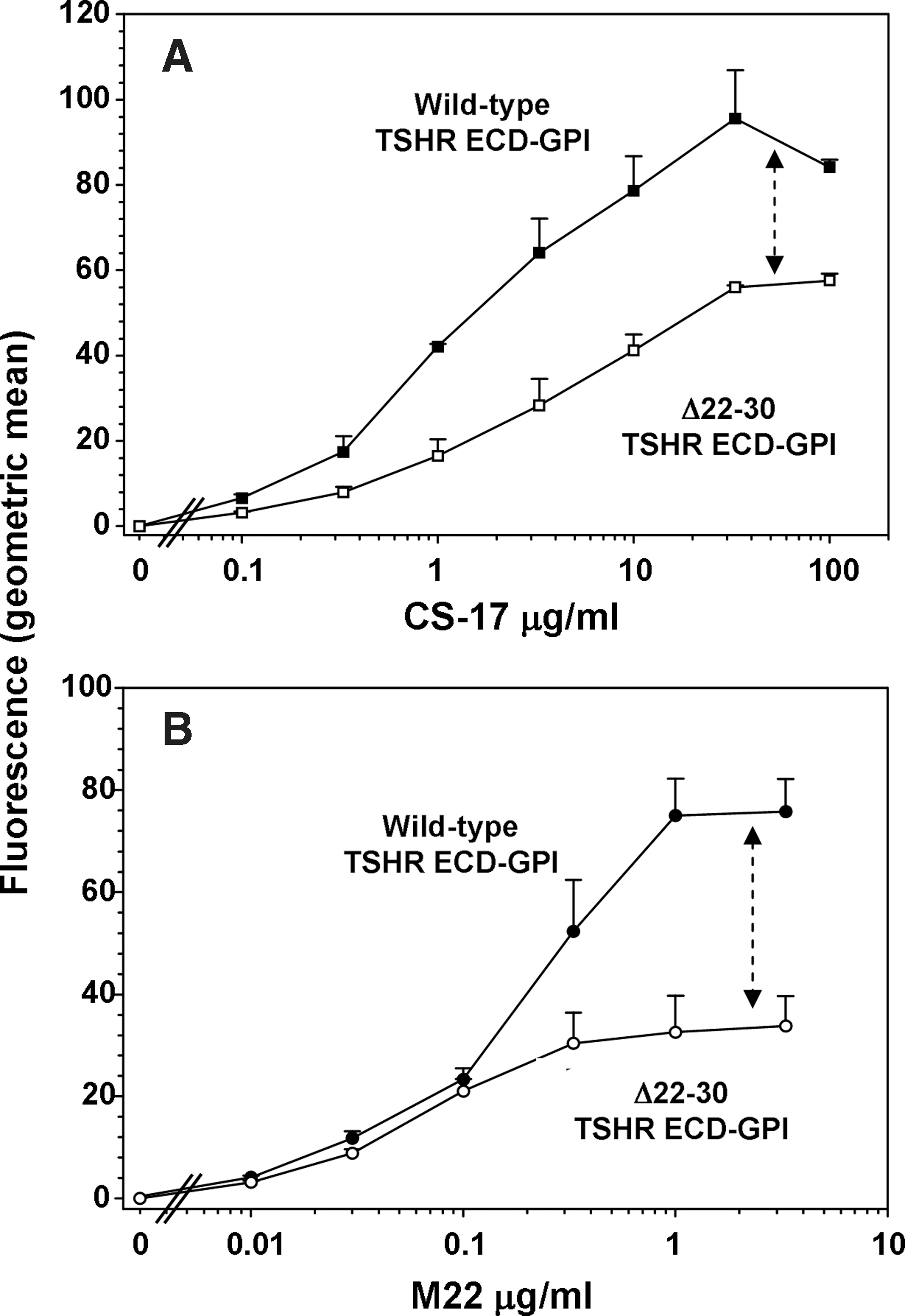
Confirmation using a second control mAb that deletion of TSHR N-terminal disulfide-linked loop 1 reduces the number of cell surface receptors detected by M22. The experiments were identical to those described in Figure 2 using CHO cells stably expressing TSHR-ECD-GPI and Δ22–30 TSHR-ECD-GPI except for the use of control murine mAb CS-17
The preceding experiments tested M22 against a control antibody (2C11 or CS-17) in parallel. In other experiments FACS was performed with M22 or 2C11 individually, but always comparing recognition of the wild-type TSHR-ECD-GPI and Δ22–30-TSHR-ECD-GPI cell lines in parallel in the same experiment. Analysis of these data supported the observation that M22 did not recognize all receptors on the cell surface. The ratios of maximum fluorescence observed with the wild-type TSHR-ECD/Δ22–30-TSHR-ECD-GPI were 3.5 ± 0.2 and 2.1 ± 0.4 (means ± SE) for M22 (10 experiments) and 2C11 (5 experiments), respectively (Fig. 4). Considering 2C11 recognition to be unaltered by the Δ22–30 deletion and, therefore, detecting 100% of cell surface receptors, M22 only detected 60.4% of these receptors. Differences in affinity between these two antibodies cannot account for this disparity because we used antibody concentrations sufficient to attain saturation of surface receptors.
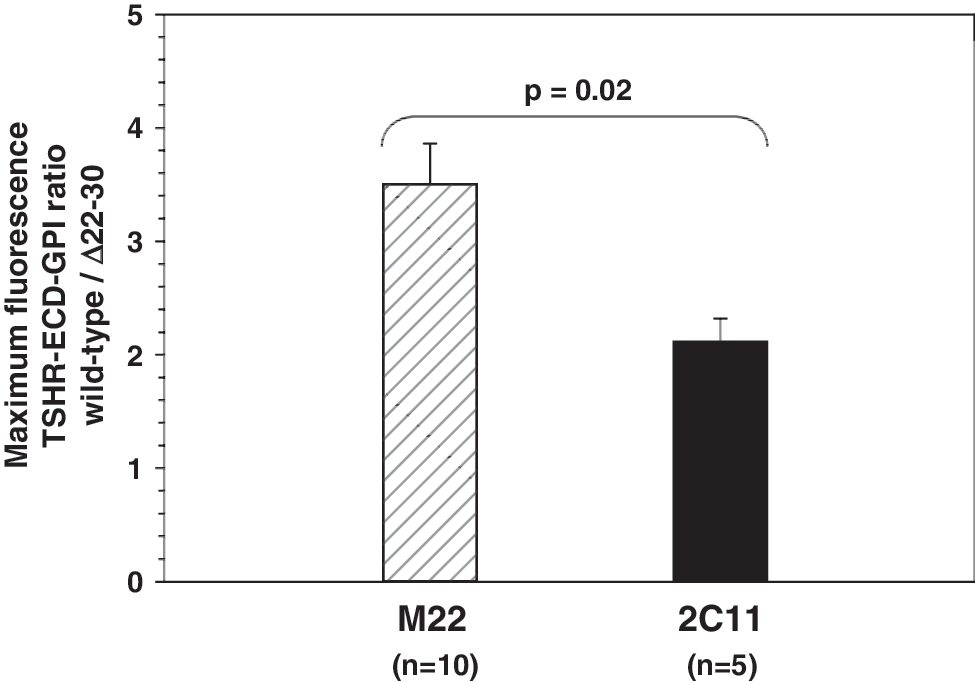
Analysis of flow cytometry experiments examining the effect of TSHR N-terminal loop 1 deletion on M22 and control antibody recognition. Although M22 and control antibodies were not always tested together (as in Figs. 2 and 3), in all experiments each antibody was tested in parallel with stably expressed wild-type TSHR-ECD-GPI and Δ22–30-TSHR-ECD-GPI cell lines. For inter-experiment comparison, the ratio of maximum fluorescence (mean ± SE) with each cell line is shown for control antibody 2C11 and for M22 (mean ± SE for 5 experiments and 10 experiments, respectively; p = 0.02, Student's t-test).
Functional studies with the TSH holoreceptor
Lacking the membrane-spanning domain, the TSHR-ECD-GPI cannot be used for functional studies. We, therefore, compared M22 stimulation of cAMP generation using the stably transfected cell lines expressing the TSH holoreceptor and this receptor with deletion of N-terminal residues 22–30. As an internal control, we used TSH because mAb 2C11 and CS-17 do not activate the TSHR and deletion of residues 22–30 does not affect the TSH binding site (7) or the functional response to TSH stimulation (17).
TSH stimulated TSHR Δ22–30 with a maximum cAMP response less than that attained with the wild-type TSH holoreceptor (Fig. 5A), consistent with a lower level of cell surface expression of the deletion mutant compared to the wild-type receptor (Fig. 2A, B). M22 elicited a proportionately similar difference in the maximum cAMP response with the wild-type TSH holoreceptor and TSHR Δ22–30 (Fig. 5B). Quantitatively, the ratios in the maximum cAMP responses for the wild-type TSH and TSHR Δ22–30 were 1.55:1 for TSH and 1.74:1 for M22, not significantly different. Therefore, although M22 recognized fewer N-terminal deletion mutations when expressed on a GPI-anchored ectodomain, with the holoreceptor the functional cAMP response to M22 was not significantly altered by the N-terminal deletion mutation.
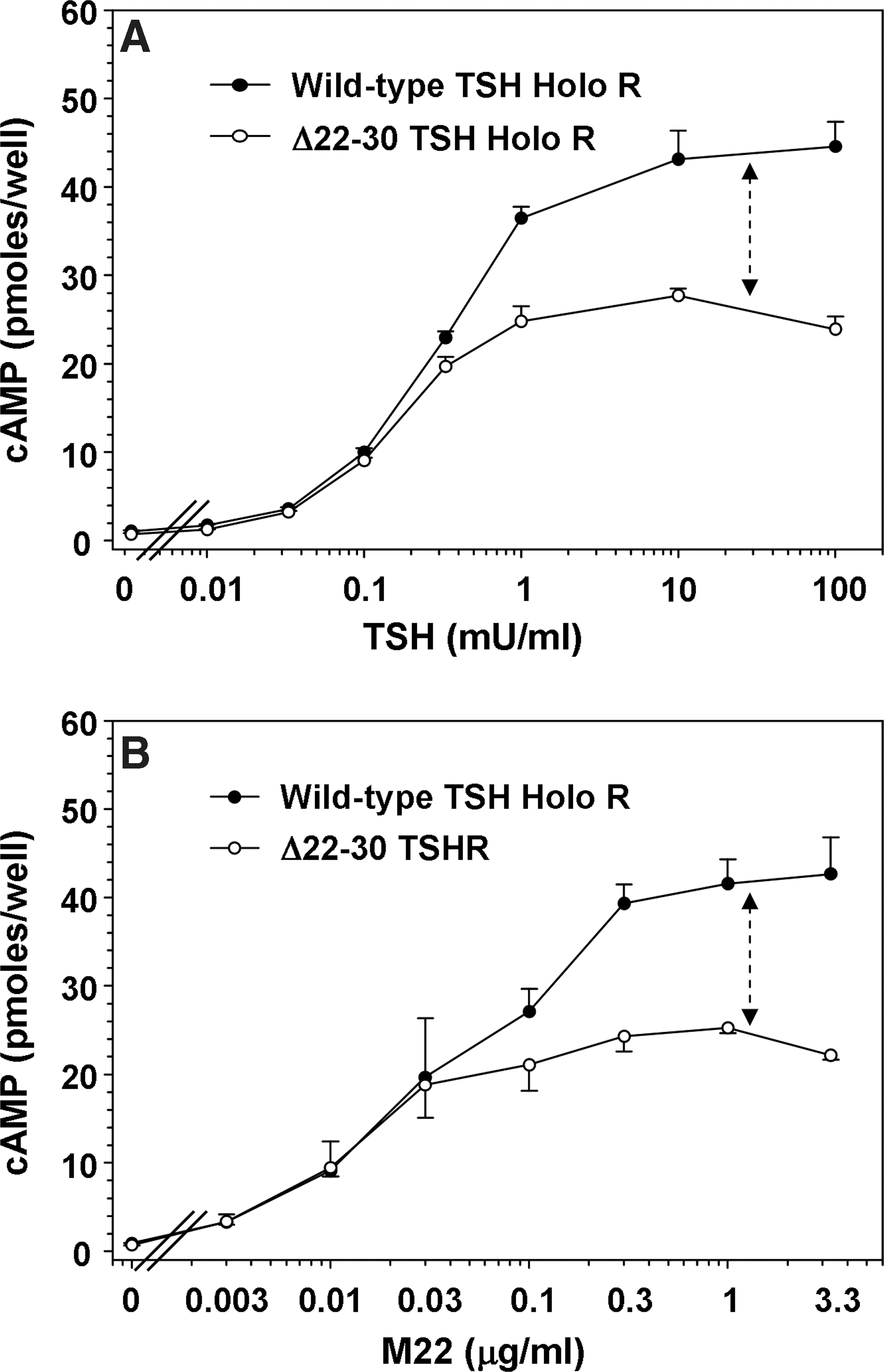
Deletion of TSHR N-terminal disulfide-linked loop 1 does not affect the cAMP response to M22 stimulation in TSH holoreceptors.
Discussion
The role of the highly immunogenic cysteine-rich N-terminus of the TSHR in TSAb activation of the receptor is controversial. Despite extensive prior evidence that the conformation of this region influences TSAb binding and function (13 –17), the crystal structure of M22 with the TSHR “showed that there were no M22 interactions involving the extreme N-terminal region” (11). For reasons outlined below, we suggest that these apparently opposing sets of information can be reconciled.
The first point to be made, even before considering our data, is that the crystal structure of the monoclonal TSAb M22 Fab with the TSHR LRD reveals two hydrogen bonds (distances of 2.93 and 3.24 angstroms) between the M22 heavy chain and TSHR residue R38 (10). The latter residues lie within the LRD N-terminal cluster of four cysteine residues (C24, C29, C31, and C41; residues 1–22 being the deleted signal peptide) (Fig. 1). Therefore, there is no doubt that TSAb does indeed interact with the cysteine-rich TSHR N-terminus.
The second point relates to ambiguity in the conformation of the free-standing TSHR LRD structure (amino acid residues 22–260) lacking the downstream hinge region and membrane-spanning domain. The crystal structure of the LRD complexed with monoclonal TSAb M22 revealed that the two disulfide bonds at the TSHR LRD N-terminus created two loops rather than the sushi domain in the closely related FSHR (Fig. 1). However, in this structure, the LRD N-terminal loop 1 (amino acid residues 22–29) could not be determined because of molecular disorder (10). These findings indicate that LRD loop 1 in this particular crystal lacks structural uniformity or has increased mobility. On the basis of these two findings, we hypothesized that TSHR LRD loop 1 was flexible around a stalk of amino acid residue E30. Our present data support this hypothesis. As revealed by the deletion of loop 1 in the TSHR N-terminal domain, M22 does not recognize all the TSHR ECD molecules tethered to the plasma membrane by a GPI anchor. The corollary of this observation is that there is, indeed, conformational heterogeneity in the TSHR ECD, at least when attached to the plasma membrane by a GPI anchor, as also occurs with the isolated TSHR LRD protein (16). Of note, the crystal structure also contains the isolated TSHR LRD. Interestingly, unlike with the TSHR-ECD-GPI, M22 does not identify different conformational forms when the identical ECD is attached to the transmembrane domain (TMD) in the TSH holoreceptor, consistent with previous data using polyclonal TSAb in Graves' patients' sera (17).
At the completion of our study, the crystal structure of the identical TSHR LRD in complex with TSHR autoantibody K1-70 (11) became available (March 2011). Remarkably, in this crystal, the structure of LRD loop 1 could be solved, confirming the N-terminal disulfide bonding deduced from the more limited information extracted from the TSHR LRD-M22 crystal. We have compared the structures of the same TSHR LRD complexed with either M22 or K1-70 by applying a colorimetric scale for the degree of structural certainty (blue) or uncertainty (red) in the macromolecular visualization tool FirstGlance in Jmol (
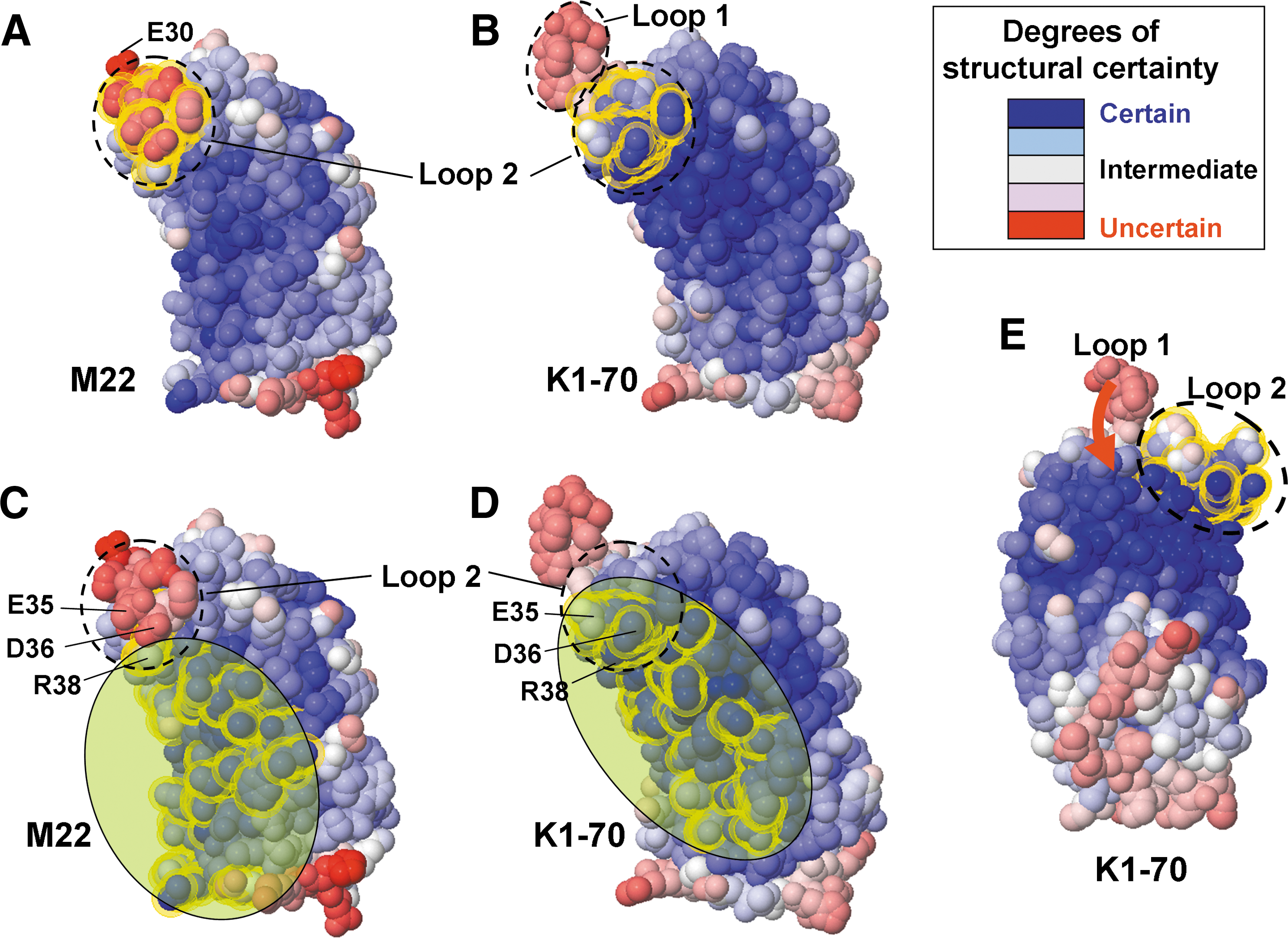
Crystal structure of the TSHR LRD visualized with a colorimetric scale for the degree of relative disorder or uncertainty. FirstGlance in Jmol (
We suggest that these differences in structural certainty between the two LRD structures can be explained by the lesser N-terminal extension of the M22 epitope (10) compared to that for K1-70 (11) (shown schematically in Fig. 6C and D, respectively). More extensive contact by K1-70 than M22 in the N-terminal cysteine cluster (contacts with residues E35 and D36 as well as R38) can have two consequences: (i) greater stabilization of LRD loop 2 (now blue vs. pink) and (ii) reduced mobility of LRD loop 1, now visualized (though pink) versus absent. The potential for LRD loop 1 mobility is evident from a more lateral view (Fig. 6E, red arrow). Indeed (more easily seen by visually rotating the molecule), forward movement of loop 1 could impinge on loop 2, thereby introducing ambiguity in the crystal structure of the latter.
The foregoing structural interpretations are consistent with our present data that M22 does not recognize all forms of TSHR-ECD-GPI present on the cell surface. In the LRD-M22 crystal relative to the K1-70 crystal, disorder of LRD loop 1 (either static or dynamic and sufficient to cause nonvisualization) is associated with greater loop 2 instability. Therefore, even though the M22 epitope does not include loop 1, the conformation or position of loop 2 may be altered by a change in the position of loop 1. With LRD N-terminal heterogeneity, M22 may not see all conformational variants.
Lacking the TMD, the TSHR-ECD-GPI is unable to signal and we therefore examined the effect of loop 1 deletion on TSH holoreceptor function, using TSH as a functional internal standard. Remarkably, loop 1 deletion was without effect on M22 functional activity. We can suggest two possible explanations for this observation. First, unlike with the TSHR-ECD-GPI, there is no apparent N-terminal heterogeneity in the holoreceptor, possibly because the two ECD forms differ in their folding or trafficking to the cell surface. If true, this alternative indicates a limitation in interpreting the crystal structure of the isolated TSHR LRD, whose structure is more closely related to the TSHR-ECD-GPI than to the holoreceptor (23). A second possibility relates to the observation that interaction of TSAb (including M22) with the TSH holoreceptor is sterically hindered (23), with a reduced binding affinity (14) compared to the TSHR-ECD-GPI (14,23). Such steric hindrance may alter the angle at which TSAb initially interacts with the ECD and mitigate, for example, interference by a particular loop 1 position.
The major accomplishment in determining the crystal structure of the TSHR LRD also provides insight into the why chimeric substitution (chimeric receptor 6-A1) (13) but not deletion of loop 1 (17) (present study) alters TSAb activity. The inter-cysteine segment in TSHR loop 1 contains two proline residues compared to only one proline in the rat LH receptor substitution (13) and none in the FSHR (Fig. 1). Prolines contribute to rigid turns in protein folding. We suggest that additional TSHR prolines permit a tight turn in loop 1, thereby permitting cysteine 1–2 and 3–4 disulfide linkage rather than a sushi domain (Fig. 1). Unlike deletion of TSHR loop 1, the presence of an N-terminal sushi domain (rather than two separate loops) would greatly alter the conformation in this component of the TSAb (including M22) epitope.
In summary, the present data support the concept that TSHR-stimulating autoantibodies interact with the cysteine cluster region at the extreme N-terminus of the TSHR LRD. Analysis of two different crystal structures of the TSHR LRD reveals structural heterogeneity and helps reconcile two apparently contradictory viewpoints. A difference between the interaction of monoclonal TSAb M22 with the identical TSHR N-terminus expressed on the TSHR-ECD-GPI versus the TSH holoreceptor suggests that crystallization of the TSHR LRD-M22 complex may not provide a complete understanding of the functional TSAb epitope(s) in Graves' disease.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health Grant DK 19289. We are also grateful for contributions by Dr. Boris Catz, Los Angeles.
Disclosure Statement
The authors declare that no competing financial interests exist.