
Abstract
Background:
Papillary thyroid carcinoma (PTC) is the most common type of thyroid cancer, whereas mucosa-associated lymphoid tissue (MALT) lymphoma of the thyroid gland is uncommon. Simultaneous occurrence of both disease entities is very rare.
Patient findings:
A 59-year-old man with known hypothyroidism from Hashimoto's thyroiditis (HT) was seen for thyroid nodules. A thyroid ultrasound revealed a heterogeneous thyroid gland with two hypoechoic nodules, one in the right aspect of the isthmus measuring 2.0 cm×3.2 cm×1.7 cm and another one in the left lobe measuring 1.4 cm×1.3 cm×1.2 cm. A fine-needle aspiration (FNA) of the right-sided nodule revealed atypical epithelial cells and atypical lymphoid cells in a background of lymphocytic thyroiditis; FNA of the left-sided nodule showed findings of PTC. A total thyroidectomy was performed. Lymph node dissection was not performed. Pathology showed extranodal marginal zone B-cell lymphoma of MALT type with extreme plasmacytic differentiation in the right nodule and PTC in the left nodule (pT1b Nx Mx). Postoperatively, he underwent radioactive iodine ablation treatment. There was only minimal neck uptake on the post-treatment scan. Further work-up did not show any evidence of extrathyroidal lymphoma. Seven years after the surgery, the patient had no evidence of recurrence of either malignancy.
Summary:
PTC is the most prevalent thyroid cancer and has an excellent prognosis. Primary thyroid lymphoma is rare and accounts for <5% of all thyroid cancers. Among the primary thyroid lymphomas, MALT lymphoma tends to have a more indolent course and a better prognosis. PTC and MALT lymphoma have been associated with HT. FNA has been validated in several studies for the diagnosis of MALT lymphoma; however, distinguishing MALT lymphoma from HT remains a challenge due to their histological similarities. The treatment of MALT lymphoma remains controversial; however, surgery is generally accepted in the early-stage MALT lymphoma as was performed in the present case.
Conclusion:
Since HT is associated with PTC and MALT lymphoma, patients with HT deserve careful surveillance for both disease entities. In our patient, the management of one malignancy did not affect the management of the other, and the prognosis did not seem to be affected.
Introduction
Patient
A 59-year-old man with hypothyroidism due to HT of 10 years of duration was seen in our department because of progressive enlargement of the thyroid gland over a few months. He had mild intermittent dysphagia to solids and liquids but denied change in voice, shortness of breath, or weight loss. He was taking levothyroxine 125 mcg daily by mouth. He denied any history of radiation to the head or neck. He reported no family history of thyroid cancer. On physical examination, he was well nourished. His trachea was midline. His thyroid gland was enlarged with the right lobe being larger than the left. A firm 3.5 cm×3.5 cm nodule was palpable in the right lobe and moved with deglutition. There was no palpable cervical lymphadenopathy. The rest of the physical examination was unremarkable.
Thyroid ultrasonography revealed a heterogeneous thyroid gland (Fig. 1). Two discrete hypoechoic nodules were noted: one in the right aspect of the isthmus measuring 2.0 cm×3.2 cm×1.7 cm and another one in the cranio-lateral aspect of the left lobe measuring 1.4 cm×1.3 cm×1.2 cm with speckled calcifications. Subsequent fine-needle aspiration (FNA) of both nodules was performed at an outside institution and submitted for our review. The right-sided nodule showed a predominance of lymphoid cells associated with rare scattered atypical epithelial cells. The lymphoid cells consisted of a relatively monotonous sample of intermediate-sized plasmacytoid lymphocytes/plasma cells. Germinal center fragments with tingible body macrophages were not seen. While this aspirate sample raised the possibility of a low-grade lymphoproliferative disorder, a definitive diagnosis could not be made due to the lack of available material for ancillary testing such as flow cytometry or immunohistochemistry. Importantly, sheets of large malignant lymphoid cells, characteristic of diffuse large B-cell lymphoma, were not seen. The atypical epithelial cells in this nodule were considered as likely representing reactive Hurthle cells. The aspirate from the left-sided nodule was distinctly different and showed a cellular sample of epithelial cells arranged in syncytial groups and microfollicles. Lymphoid cells were inconspicuous. Nuclei were enlarged, oval, and overlapping. Diagnostic nuclear features of PTC, including pale, powdery chromatin, nuclear grooves, small nucleoli, and intranuclear inclusions, were seen.
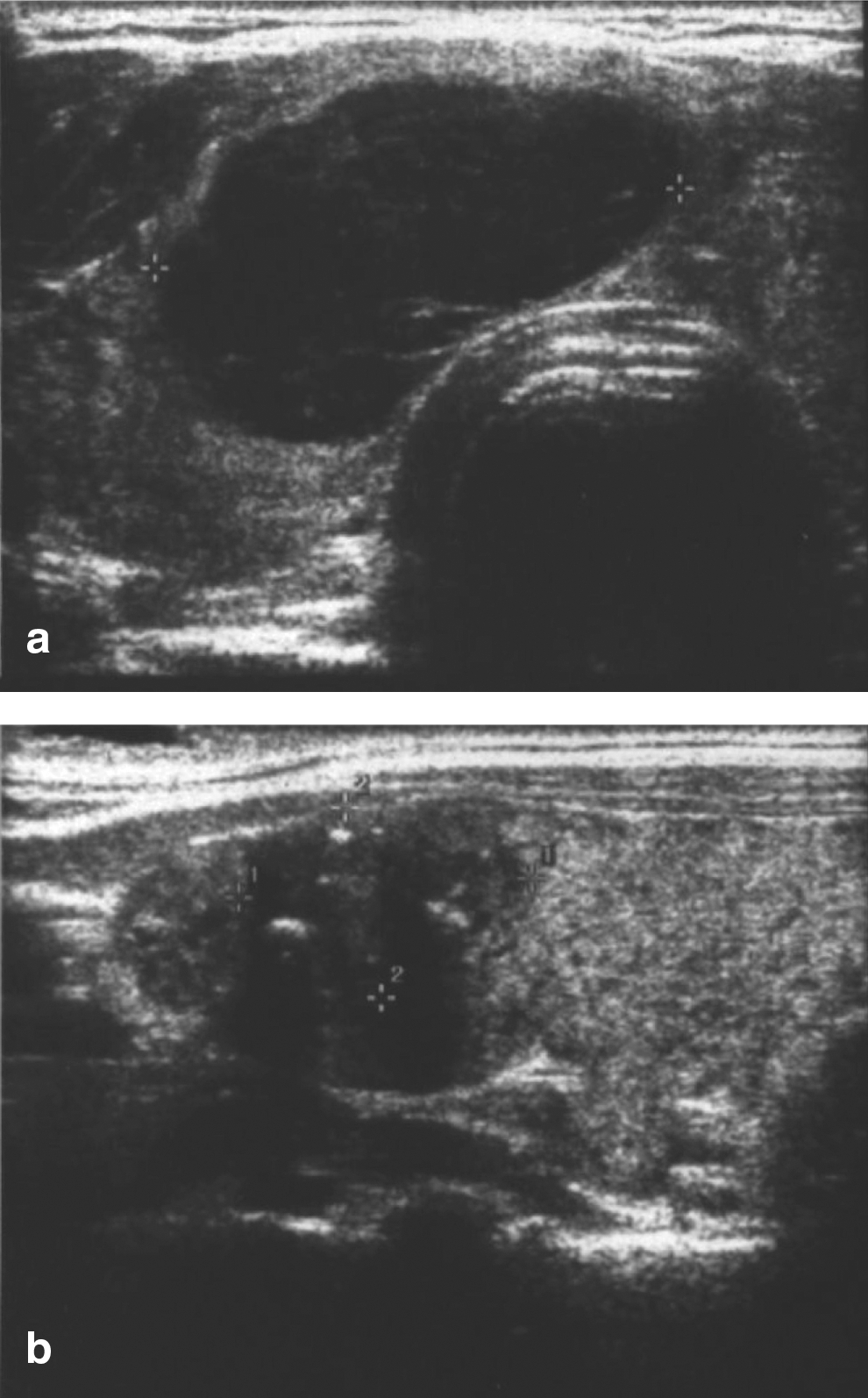
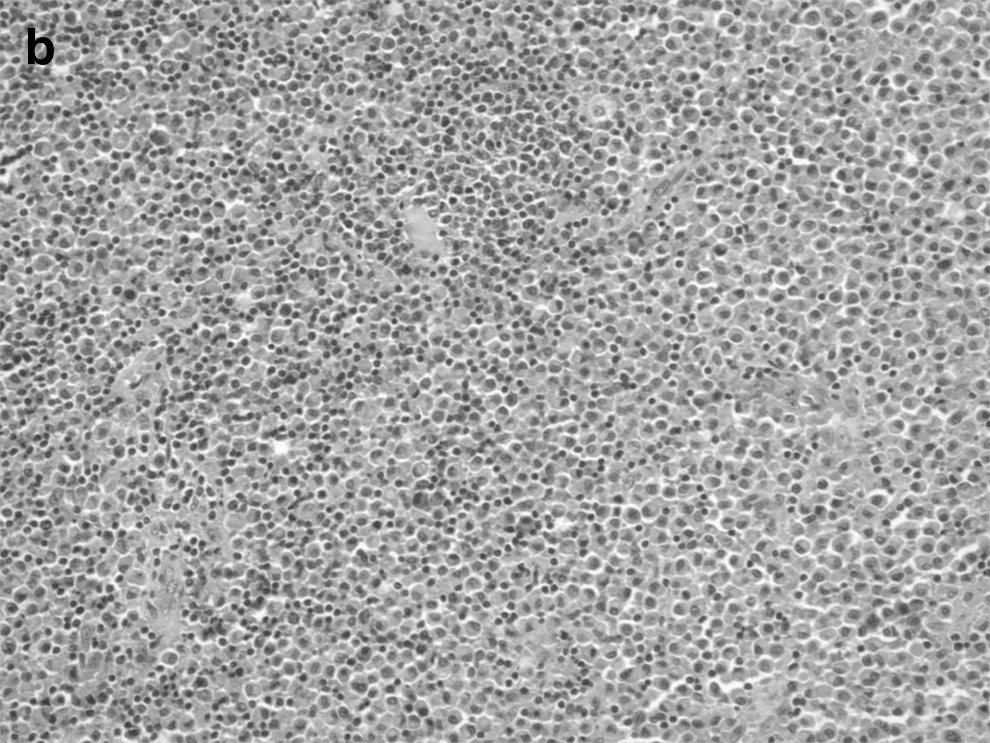
A total thyroidectomy was performed. No lymph nodes were removed. Histopathology revealed an extranodal marginal zone B-cell lymphoma of MALT type with extreme plasmacytic differentiation in the right nodule (Fig. 2). The MALT lymphoma had a nodular architecture with nodules comprised nearly exclusively of plasmacytoid cells. Scattered reactive germinal centers were also seen. Rare lymphoepithelial lesions were present at the periphery of the plasmacytic nodules. Immunohistochemical staining was performed, and the plasmacytic cells were kappa light-chain restricted. The histology from the left lobe showed the PTC as possessing a predominantly follicular pattern. The tumor measured 1.8 cm×1.8 cm×1.5 cm, and there was no extrathyroidal extension (Fig. 3).

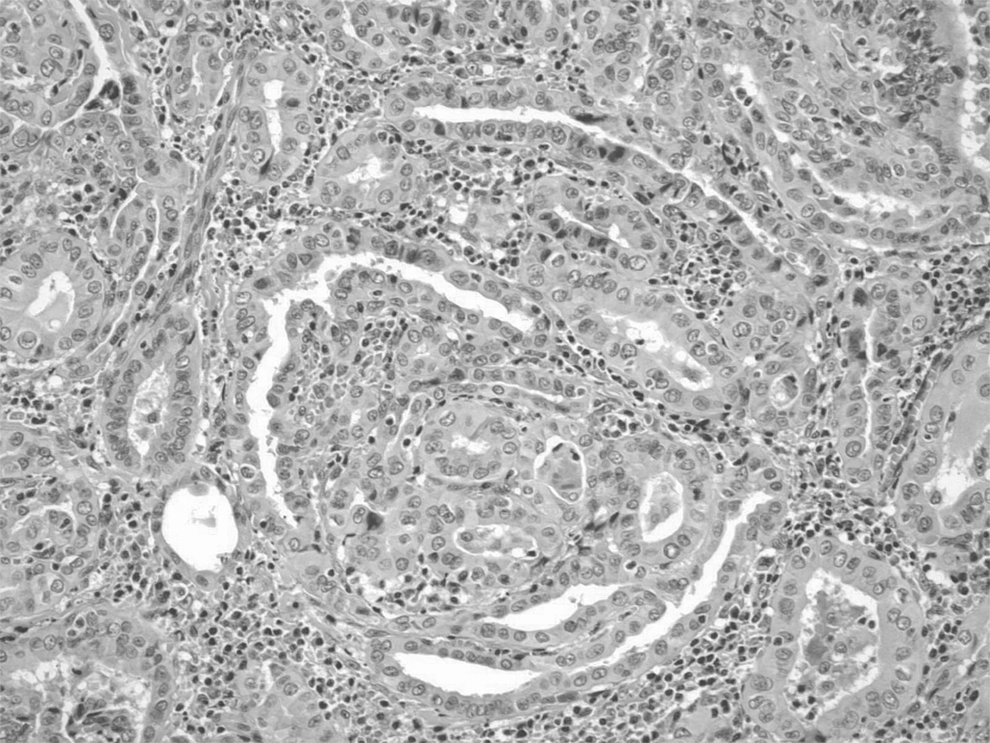
Papillary thyroid carcinoma with a predominant follicular pattern and diagnostic nuclear features (20× magnification).
The patient was discharged home on levothyroxine 125 mcg. Two months later, he underwent remnant ablation with 75 mCi (2.78 GBq) of 131I after withdrawal from the thyroid hormone. A whole-body scan performed after the 131I treatment did not reveal any areas of abnormal iodide uptake. Bone marrow aspiration biopsy was normal. Computed tomography scan of the chest, abdomen, and pelvis did not show any evidence of lymphoma. There was no evidence of disease on his last visit, that is, 6 years after the thyroid surgery.
Discussion
PTC and MALT lymphoma have been associated with HT. Chronic antigenic stimulation in HT has been postulated as leading to the development of malignancy (2). In addition, studies have suggested that overlapping molecular mechanisms between HT and PTC such as RET/PTC gene rearrangement may regulate the early stages of tumor development and inflammation of the thyroid (4,5). PTC has been reported as being associated with HT in 10%–58% of cases (1,4); whereas HT has been found in more than 90% of reported cases of thyroid lymphoma (2). The development of lymphoma has been estimated as being 40–80 times higher in patients with chronic thyroiditis compared with the general population (6). The coexisting occurrence of PTC and thyroid lymphoma is rare. To our knowledge, only one case has been reported (3). The patient, a 61-year-old man, presented with a rapid enlargement of the thyroid gland over a 3-month period. Pathology showed multicentric PTC, accompanied by HT and a concomitant thyroid MALT lymphoma. There was no evidence of other tissue involvement by the lymphoma after a staging work-up. He underwent radioactive iodine therapy and had a negative whole-body scan. He had no evidence of recurrence after 6 years of follow-up.
PTC is the most common type of thyroid cancer, and it has a 20-year survival of >90%. PTC has distinct cytologic features of sheets, papillary architecture, and/or microfollicles with characteristic nuclear features such as nuclear enlargement, pale powdery chromatin, grooves, small but distinct nucleoli, and intranuclear cytoplasmic inclusions (7). Hypoechogenecity and the presence of microcalcifications on ultrasonography increase the suspicion of PTC (8); these features were noted in the left sided nodule and correlated with histological findings. Other typical findings include irregular shape, poorly defined borders, and intranodal vascularity on Doppler ultrasonography. Surgery is the mainstay of treatment and is often curative.
Among the primary thyroid lymphomas, MALT lymphoma tends to have a more indolent course and carries a better prognosis with a 5-year disease-specific survival rate >90% (2). The diagnosis is based on the presence of a combination of morphologic features and immunophenotyping by flow cytometry and immunohistochemistry. Diagnostic features of MALT lymphoma include lymphoepithelial lesions, reactivation germinal centers, and frequent plasmacytic differentiation (9). The diagnosis of MALT lymphoma is difficult in the setting of HT, as both disease entities have similar histologic characteristics, including infiltration by B-cells, plasma cell differentiation, and lymphoepithelial lesions (10). Thus, careful attention toward the histology and ultrasonographic findings is important. In addition, it is important to collect material for ancillary testing at the time of FNA in patients suspected of having a thyroid lymphoma. This is especially true for patients with MALT lymphoma, given the morphologic overlap with HT in both cytologic preparations. While diagnosis of diffuse large B-cell lymphoma by FNA is often straightforward, the diagnosis of MALT lymphoma requires immunophenotyping. Ideally, at least one dedicated pass in a cell-preservative solution such as Roswell Park Medical Institution media will allow for immunophenotyping by flow cytometry. Alternatively, cytology cell blocks can be prepared from the needle rinses for immunohistochemical staining. On-site assessment of specimen adequacy by cytologists at the time of FNA allows the immediate triage of aspirate samples for ancillary testing. The presence of a highly cellular lymphoid specimen with intermediate-sized cells of minimal atypia and a few epithelial-follicle cells in the absence of tingible body macrophages is suspicious for low-grade MALT lymphoma in the setting of HT (11). The finding of sheets of cells with abundant basophilic cytoplasm but eccentric lymphocyte-like nuclei helps differentiate MALT lymphoma with extreme plasmacytic differentiation from HT with reactive plasma cell infiltrates (12). B-cell lymphomas are monoclonal and demonstrate restricted expression of either lambda or kappa light chains (13). In our case, in addition to histologic features characteristic of MALT lymphoma, monoclonality was demonstrated in the thyroidectomy specimen by proving that the plasma cells present showed kappa light chain restriction. A reactive plasma cell infiltrate, as is often seen in HT, would be comprised of polyclonal plasma cells expressing both kappa and lambda light chains. In a case series by Sangalli et al., the combination of morphologic criteria and immunocytochemical staining resulted in the differentiation of lymphoma from HT in 70% of the cases (11). Primary thyroid lymphoma appears as a well-delineated, markedly hypoechoic nodule with enhancement of the posterior wall on ultrasonography, and calcifications are not usually seen (14).
The treatment of primary thyroid lymphoma depends on staging according to the Ann Arbor classification for lymphomas. Pure MALT lymphoma is usually localized to the thyroid gland (Stage 1E) and responds well to local therapy such as total thyroidectomy or radiation therapy alone (2). About 40% of diffuse B-cell lymphoma appears to evolve from MALT lymphoma; thus, early diagnosis is prudent (2). The overall 5-year survival is <50%. Treatment of this subtype includes chemotherapy and radiation.
There has been no literature to date regarding the optimal management of the simultaneous occurrence of PTC and MALT lymphoma. In the author's opinion, the management of one is not affected by the management of the other, and the prognosis is probably affected by the one having the worst stage.
Conclusion
Patients with HT should receive careful surveillance for both PTC and MALT. They should be monitored for new neck symptoms and palpable abnormalities. If nodules are found, then the possibility of PTC or MALT should be considered. Our patient with concomitant PTC and MALT in the setting of HT is doing well 6 years after thyroidectomy and 131I treatment; however, there is little published information on what the overall prognosis of a group of such patients is likely to be.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.