
Abstract
Background:
Three endocrine neoplasms—bilateral pheochromocytomas, somatotrophic pituitary adenoma inducing acromegaly, and papillary carcinoma of the thyroid—occurred concurrently in a patient. A genetic mutation was hypothesized. Possible previously described genetic mutations were explored.
Methods:
Clinical assessments, laboratory data, images of tumors, histopathology, and immunohistochemistry of excised tissues documented the three neoplasms. Clinical assessment of the patient, family history, and a review of the literature sought a familial basis for the disorders.
Results:
The methods confirmed the presence of three endocrine neoplasms. Each neoplasm was surgically excised and histologically verified. Surgical and 131I treatments reduced the papillary carcinoma, but eventually this tumor progressed to a lethal degree. History, including that of nine siblings, uncovered no familial neoplasms. No similar case was found in the literature, but possible associations with germline mutations were considered.
Conclusions:
The concurrent development of pheochromocytomas, pituitary somatotrophic adenoma, and papillary thyroid carcinoma appears to be unique. Nevertheless, such tumors, particularly bilateral pheochromocytomas, strongly suggest a de novo germline mutation in a gene not previously associated with multiple endocrine neoplasia syndromes.
Introduction
The features of MEN-2, medullary carcinoma of the thyroid (MTC), pheochromocytomas, and hyperparathyroidism derived from parathyroid gland hyperplasia, were first described in 1961 (3), but it soon became clear that a subset of patients exhibited MTC, pheochromoctyomas, and pronounced facial features from ganglioneuromas, but not hyperparathyroidism (4). The former combination of familial tumors was designated MEN-2A and the latter, MEN-2B. Germline mutations of the RET protooncogene underlie these multiple endocrine disorders (5).
Variations in the specific mutations of oncogenes have generated diverse phenotypic appearances in affected individuals. However, none have exhibited the combination of the three endocrine neoplasias—bilateral pheochromocytomas, somatotrophic pituitary adenoma, and papillary carcinoma of the thyroid—manifested in our patient.
Case Presentation
When initially evaluated at the University of Michigan Health System in December 1996, the patient was a 29-year-old male carpenter. For about 2 years, he was afflicted by headaches and diaphoresis, both day and night. His elevated blood pressure was unmitigated by medications, including atenolol 25 mg/day. Yet, he continued to work regularly. In November 1996, a search for a cause of secondary hypertension included a CT that portrayed bilateral adrenal enlargements attributable to pheochromocytomas (Fig. 1A, B). He had no visual symptoms. Libido and erectile function were reported to be normal.
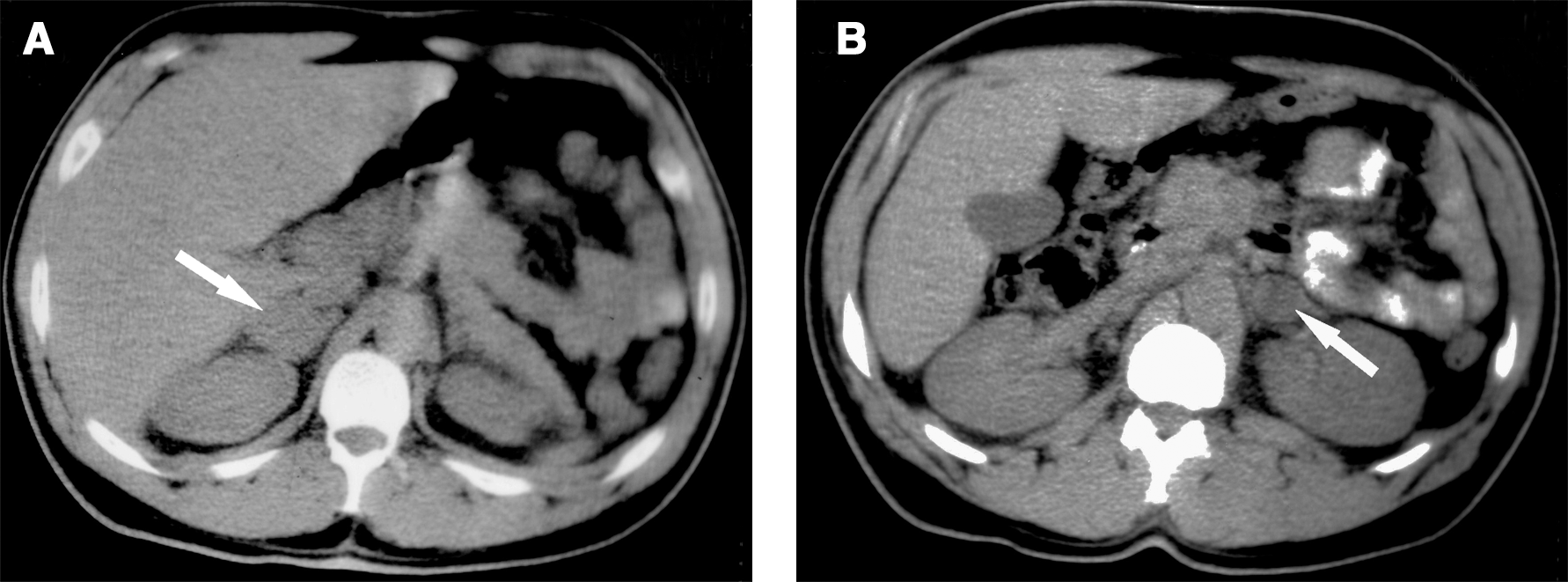
CT images of abdomen at adrenal gland levels, November 1996. Arrows point to the depicted abnormalities:
He weighed 118 kg and stood 188 cm. Blood pressure was 170/116 and heart rate was 72 beats/min. Soft tissue swelling was prominent in his large hands and feet. His shoe size was 13 but was recently unchanged. Thickened skin was damp from perspiration. A number of small skin lesions were observed; one appeared to be a lipoma, but none were characteristic of a myxoma or a neurofibroma. Readily apparent were a large brow and an overbite in a prominent jaw. No abnormality was detected in either his visual fields or extraocular muscle movements. His tongue was expansive. Palpable enlargement of his left thyroid lobe was accompanied by two easily discerned lymph nodes in his left neck.
He was never married and had no children. Nine siblings were described: six sisters were 2–19 years older, and three brothers were 4–18 years older. None had developed the stigmata observed in the patient. Although one brother was said to be large, a family picture showed none of the siblings to be as tall as the patient. A sister died at age 45 years of a “heart problem.” The eldest sister had been diagnosed with heart disease. His father developed Hodgkin's disease and prostate cancer; he died at age 72. His mother died of metastatic uterine cancer at age 69. Maternal and paternal grandparents, and uncles and aunts lived to old ages. Nieces and nephews were reported to be in good health. The family heritage was Polish, German, and Bahamian.
Analysis of urine showed elevated excretion rates of epinephrine (87 μg/24 hours; normal ≤20), norepinephrine (1090 μg/24 hours; normal ≤100), and vanillylmandelic acid (26 mg/24 hours; normal ≤10). The serum level of insulin-like growth factor (IGF-1) was elevated at 1048 ng/mL (normal ≤380), but the growth hormone (GH) concentration was normal. Concentrations of serum calcium, phosphorus, and parathormone were normal. Serum prolactin (PRL) was subnormal (2.0 ng/mL; normal 3.0–23.0), as was serum testosterone (2.0 ng/mL; normal 2.5–9.5).
MRI images of the sella turcica depicted a pituitary adenoma without extension into the suprasellar space or the cavernous sinus (Fig. 2); the floor of the sella was not optimally visualized.
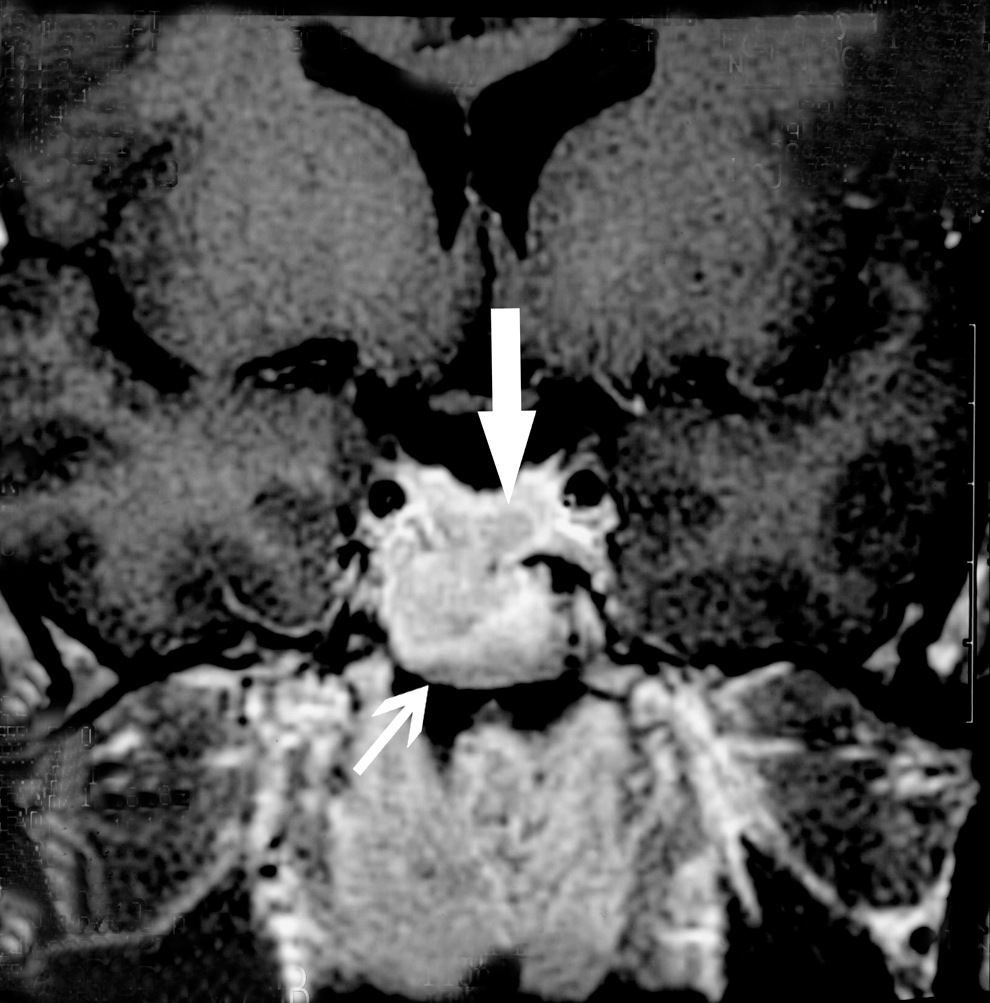
Gadolinium-enhanced MRI image of sella turcica in December 1996 demonstrates a central region of diminished enhancement within the gland attributable to a pituitary adenoma (thick arrow); immediately inferior to the region is a large mucosal retention cyst within the sphenoid sinus (thin arrow).
During phenoxybenzamine therapy on January 22, 1997, the pheochromocytomas were excised by bilateral total adrenalectomies. The surgeon reported that “there did not appear to be any normal adrenal gland remaining” on the right side. The pathologist recorded complete adrenalectomies; the right adrenal gland was the larger and contained two pheochromocytomas, while the left gland contained a pheochromocytoma as well as nodular medullary hyperplasia (Fig. 3A, B). Bilateral pheochromocytomas with medullary hyperplasia are typical of those observed in familial cases.
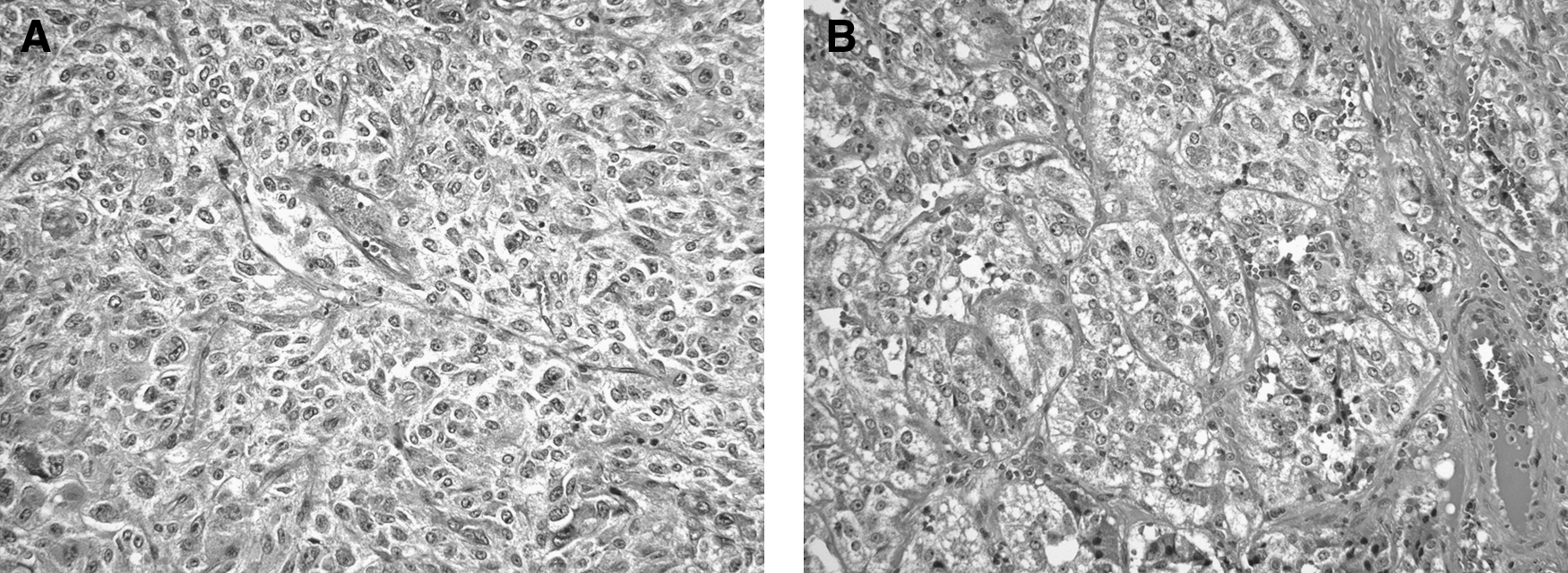
Histopathology of bilateral and multifocal pheochromocytomas.
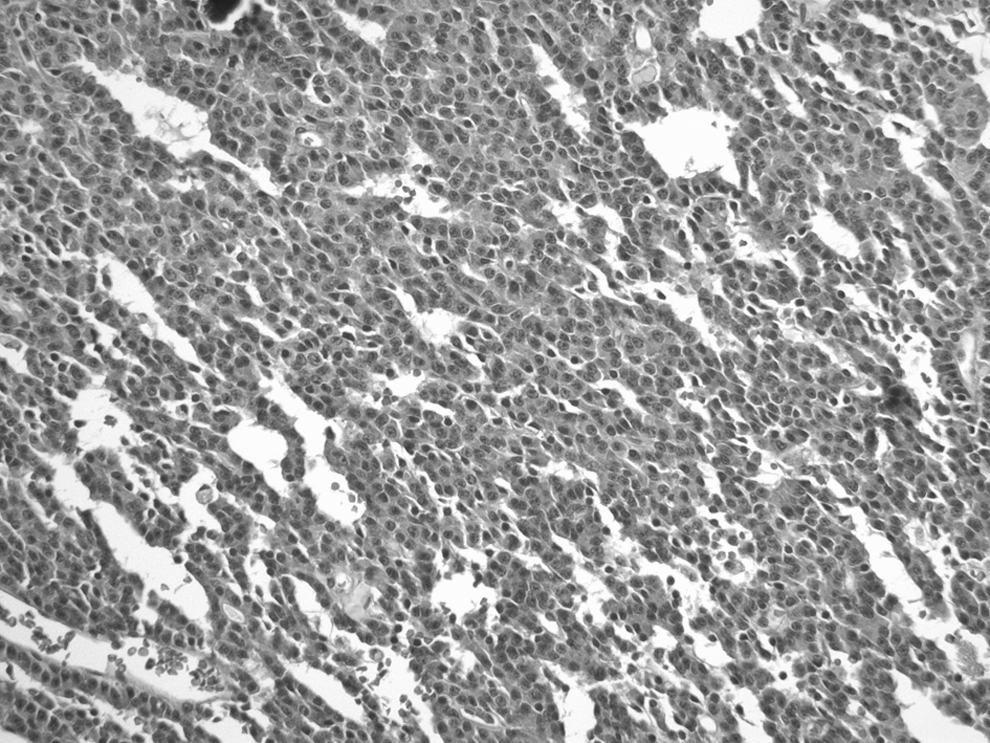
A prior ultrasound-guided fine needle biopsy enabled aspiration of papillary carcinoma cells from the left thyroid nodule, and, thus, under the same anesthesia, a total thyroidectomy removed a nodular left lobe and normal-appearing right lobe, isthmus, and pyramidal lobe. Multiple enlarged nodes were dissected free from the left neck. Histologic examinations displayed extensive primary papillary carcinoma that replaced most of the left lobe and also involved the right lobe. The majority of the tumor displayed a follicular growth pattern, but areas of papillary architecture were present (Fig. 4A, B). The tumor was invasive and extended into extrathyroidal tissues. Numerous cervical lymph nodes contained metastatic papillary carcinoma. Postoperative daily maintenance therapy consisted of hydrocortisone in divided doses, 15, 10, and 5 mg; fludrocortisone 0.1 mg; and thyroxine 0.2 mg.
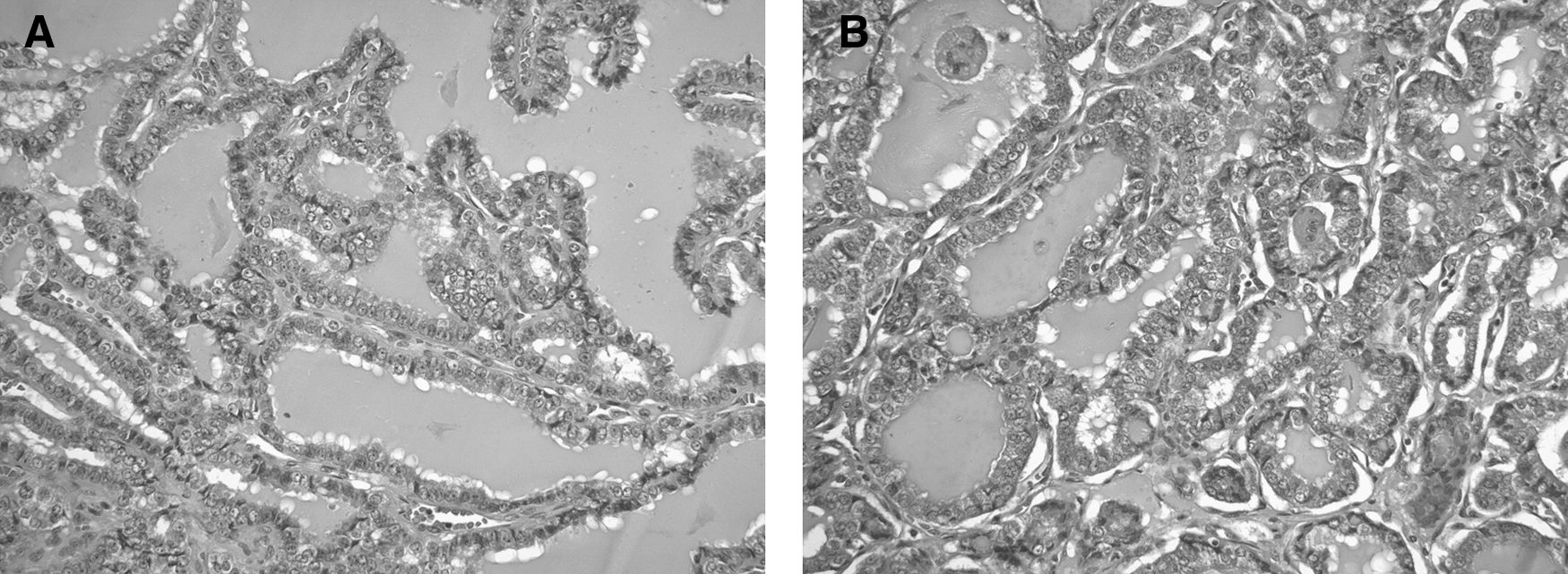
Primary and metastatic papillary thyroid carcinoma.
In March 1997, thyroxine therapy was temporarily withheld, and serum thyrotropin (TSH) rose to 51 mU/mL (normal 0.3–5.0), and thyroglobulin was 74 ng/mL. He was treated with 7.4 GBq (200 mCi) of 131I, and 2 days later whole body scintiscans revealed 3 foci of radioactivity in his left neck, and smaller foci in his right neck (Fig. 5A). Over the subsequent years his thyroxine dose was changed in efforts, not always successful, to suppress his TSH below 0.1 mU/L.
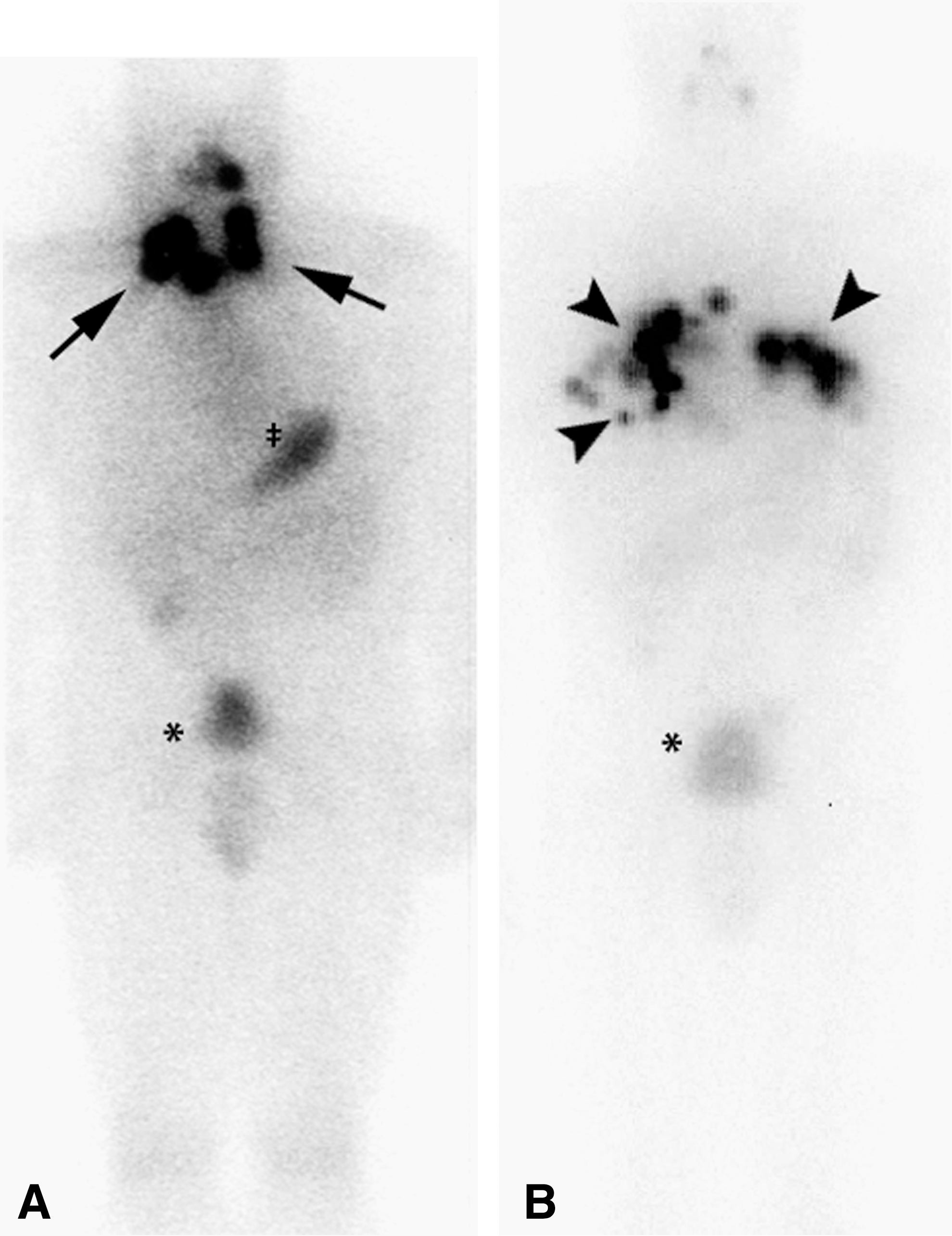
Scintigraphic images displaying 131I concentrations.
On March 21, 1997, a transphenoidal resection appeared to have completely removed his pituitary adenoma. The tumor was strongly positive for chromogranin A and focally positive for growth hormone (Fig. 6). The postoperative value for IGF-1 was lower but still elevated at 506 ng/mL. Medications were unchanged at this discharge from the hospital. Re-evaluation on April 30, 1997, recorded his blood pressure as 150/80 sitting and standing, and he weighed 120 kg.
Pituitary adenoma. The tumor displayed a typical growth pattern of a well-differentiated neuroendocrine neoplasm with small round nuclei and abundant eosinophilic cytoplasm. Focally, the tumor contained numerous psammomatous calcifications. By immunohistochemistry, the tumor cells were strongly immunoreactive to chromogranin A, focally positive for growth hormone, and negative for follicular stimulating hormone, luteinizing hormone, thyrotropin, adrenocorticotrophic hormone, and PRL.
On April 1, 1998, a scintiscan displayed foci of radioactive iodine in his neck that were less intense than in 1997, but his hypothyroid-stimulated thyroglobulin concentration was higher (191 ng/mL). A second treatment with 131I, 7.4 GBq, was administered. Subsequent care was complicated by miscommunications between patient and physicians.
While hypothyroid in June 2005, imaging with single-photon tomography of diagnostic 131I combined with CT showed lung nodules and hilar nodes containing radioactivity. He received his third treatment, another 7.4 GBq of 131I; images of this radioactivity prominently displayed the metastases (Fig. 5B).
In September and December of 2005, he felt well and was working regularly. Under thyroxine suppression, his thyroglobulin had declined to 22 ng/mL. During the latter evaluation, a CT with contrast medium demonstrated a large hilar node, but no change in small lung metastases; also portrayed was adrenal gland tissue not dissimilar from the original normal gland (Fig. 7).
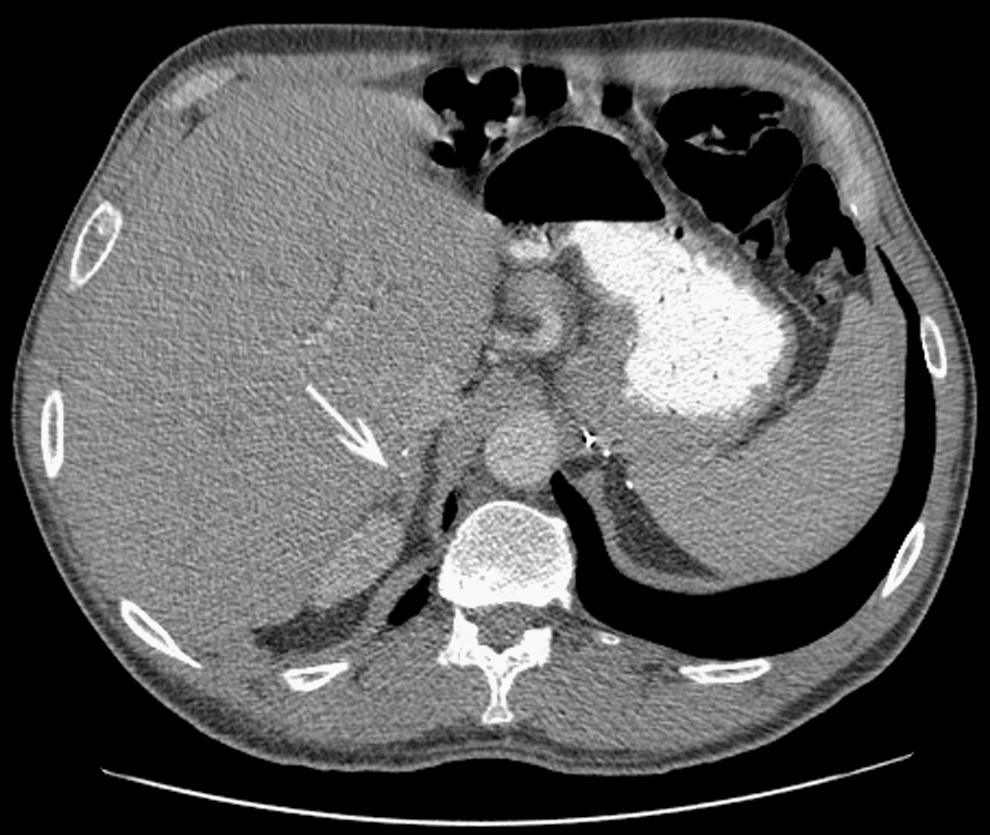
In December 2005, surveillance abdominal CT demonstrates a normal-appearing right adrenal gland (arrow); surgical clips in the right and left adrenal beds are residua of bilateral adrenalectomies.
He returned on July 5, 2007, feeling well and working regularly. However, his prescriptions for hydrocortisone and fludrocortisone had expired 2 months previously, and he was without these medications thereafter. Thyroxine was continued at 0.2 mg/day. His weight was unchanged from the previous visit; his blood pressure was 137/85 and his heart rate, in beats/min, was 40 while sitting and 54 standing. There was no increased pigmentation of his skin or oral mucus membranes. Assessments of adrenal cortex function gave plasma levels of adrenocorticotrophic hormone (ACTH), 36 pg/mL (normal 9–52), and cortisol 0.15 mg/L (normal 0.07–0.32). It was apparent that he harbored accessory adrenal cortical tissue, and hydrocortisone and fludrocortisone were not re-prescribed. While his TSH remained suppressed, his serum thyrogobulin had risen to 112 ng/mL. His IGF-1 level was then normal.
The patient was last seen on December 9, 2008. His weight was unchanged; his blood pressure was 127/67 and heart rate 49 beats/min. Again no lymph nodes were palpable in his neck, and the physical examination was normal. Plasma concentrations of ACTH and cortisol remained normal. Yet, during thyroxine therapy, his serum thyroglobulin concentration had increased further (263 ng/mL). Still, he was feeling well and working regularly.
The patient developed lower right back pain in mid-2009. In another medical center, MRI uncovered tumor in his right pelvis; biopsy demonstrated thyroid carcinoma. Despite treatment with external beam radiation and chemotherapy, the neoplasm progressed to his left pelvis, vertebrae, and ultimately to his brain. Experimental therapy was unhelpful, and he died on October 1, 2010.
Discussion
Our patient manifested a unique combination of endocrine tumors: bilateral pheochromocytomas within a background of medullary hyperplasia, pituitary adenoma secreting growth hormone and associated with acromegaly and possibly gigantism, and papillary carcinoma of the thyroid. No known syndrome or conceivable interrelationships among the tumors explained the combination. The family history was unrevealing. Nevertheless, a de novo germline mutation seemed likely.
Patients with bilateral pheochromocytomas have familial and/or germline mutations in 59 (6) and 76 (7) percent of cases, respectively. The bilateral tumors are most frequently observed in Von Hippel-Lindau (VHL) disease, MEN types 2A and 2B, and neurofibromatosis type 1 (NF1) (6,7). Bilateral pheochromocytomas are occasionally seen in patients with a succinate dehydrogenase D (SDHD) mutation (7), but have not been described in those carrying the SDHB gene. Although mutations of SDHB and SDHD are frequently associated with pheochromocytomas, paragangliomas are also common; the latter manifestation was not present in our patient.
The literature offers little evidence from which to construct further associations. Major components of VHL disease—hemangioblastoma of the central nervous system, renal cell carcinoma, pancreatic cysts, retinal angioma, and epididymal cysts (8)—were absent in our patient, but possibly the latter two features could have been overlooked. In contrast to pheochromocytomas developing in the MEN-2 syndromes, the tumors arising as part of VHL disease usually do not secrete epinephrine (9), a product which was excreted at an elevated rate in our patient.
In addition, the almost invariable component of MEN-2, MTC, was absent in our patient although a very small focus could have been overlooked. A careful examination had revealed none of the common lesions of NF1: neurofibromas, café-au-lait spots, axillary freckling, and abnormality of the iris (10).
Growth hormone–secreting pituitary adenomas have been associated with MEN type 1 (11) and the mutation of aryl hydrocarbon receptor–interacting protein gene (11 –13). A possible association within our patient appears in the report demonstrating that the frequency of papillary carcinoma of the thyroid was increased in patients with acromegaly. In 125 patients with acromegaly, 7 developed papillary carcinoma of the thyroid, 2 of which were the follicular variant; a carcinogenic role for IGF-1 was proposed (14).
Papillary carcinomas of the thyroid are components of the syndrome of phosphatase and tensin PTEN homolog mutation (15,16), Cowden syndrome, and familial adenomatous polyposis (17). However, neither pheochromocytomas nor pituitary adenomas were included in these familial associations. Nevertheless, familial non-MTCs of the papillary type were found relatively frequently to metastasize to bilateral cervical nodes and lead to a dire prognosis (18), as in our patient.
Another combination has been reported; papillary carcinoma of the thyroid was developed in a patient with bilateral pheochromocytomas associated with a paraganglioma but no mutation was found to indict VHL, MEN, SDHB, or SDHD (19).
Accessory adrenal cortical tissue has been reported following adrenalectomies (20) and has sustained Cushing's syndrome in other patients (21,22). The frequency of this anomaly is unknown, and, being nonneoplastic, the accessory adrenal cortex maybe unrelated to the three endocrine neoplasms in our patient.
We could not find a link that would associate the three endocrine tumors in our patient. Nonetheless, novel mutations are frequently reported, for example, in the MEN-1 (23) and MEN-2 (24) syndromes. It is reasonable to consider that one or more of the germline mutations discussed previously were at the base of our patient's three endocrine disorders. Unfortunately, blood was not obtained for genetic analyses before his demise.
Addendum
Since acceptance of this manuscript by the editors of Thyroid, the authors were made aware of a publication (25) describing a 37-year-old man who exhibited multiple paragangliomas, bilateral pheochromocytomas and a growth hormone–secreting pituitary adenoma associated with a novel SDHD mutation; family members displayed paragangliomas.
Referenced was an article we had overlooked (26). This study cited a previously described (27) 52-year-old woman who manifested: a right adrenal pheochromocytoma that was invasive, a chromophobe pituitary adenoma, and papillary carcinoma of the thyroid.
Conclusion
We present a patient with three concurrent endocrine neoplasms: bilateral pheochromocytomas, somatotrophic adenoma inducing acromegaly, and metastatic papillary carcinoma of the thyroid. Accessory adrenal cortical tissue was also found but maybe unrelated. Family history uncovered no relative with a comparable phenotype. Any one of a number of mutations is possible, but unfortunately blood for genetic analysis was not obtained before the patient died of progressive thyroid carcinoma. As technology to perform whole genome sequencing using paraffin-embedded tissue advances, this case will be revisited in an attempt to identify a novel gene mutation.
Footnotes
Acknowledgment
The authors are indebted to Jennifer Guess for expert help in labeling the figures.
Author Disclosure Statement
All authors declare that no competing financial interests exist.