
Abstract
Background:
We previously reported the partial effectiveness of imatinib (also known as STI571, Glivec, or Gleevec) on anaplastic thyroid cancer (ATC) cells. Imatinib is a selective tyrosine kinase inhibitor that has been used for various types of cancer treatments. Recently, several reports have demonstrated that imatinib enhanced the sensitivity of cancer cells to other anticancer drugs. In this study, therefore, we investigated whether imatinib enhances the antitumor activity of docetaxel in ATC cells.
Methods:
Two ATC cell lines, FRO and KTC-2, were treated with imatinib and/or docetaxel. Cell survival assay and flow cytometry for annexin V were used to assess the induction of apoptosis. Changes of pro- and antiapoptotic factors were determined by Western blot. Nuclear factor-κB (NF-κB) activity was measured by DNA-binding assay. Tumor growth was also investigated in vivo.
Results:
The combined treatment significantly enhanced apoptosis compared with single treatment. ATC cells themselves expressed high levels of antiapoptotic factors, X-linked inhibitor of apoptosis (XIAP), and survivin. The treatment with docetaxel alone further increased their expressions; however, the combined treatment blocked the inductions. Although imatinib alone had no effect on NF-κB background levels, combined treatment significantly suppressed the docetaxel-induced NF-κB activation. Further, the combined administration of the drugs also showed significantly greater inhibitory effect on tumor growth in mice xenograft model.
Conclusions:
Imatinib enhanced antitumor activity of docetaxel in ATC cells. Docetaxel seemed to induce both pro- and antiapoptotic signaling pathways in ATC cells, and imatinib blocked the antiapoptotic signal. Thus, docetaxel combined with imatinib emerges as an attractive strategy for the treatment of ATC.
Introduction
Docetaxel is an anticancer microtubule-stabilizing agent that induces apoptosis by suppressing the microtubule dynamics of mitotic apparatus. Docetaxel also arrests cells in G2/M phase, leading to increase of sensitivity of cancer cells to radio- and chemotherapies (2 –4). Although the effect of the drug alone was only modest against ATC in a clinical trial (5), combined therapy with radiation and/or another type of drug may be effective (6 –8).
Imatinib (also known as STI571, Glivec, or Gleevec) is a selective tyrosine kinase inhibitor and was originally developed to inhibit BCR/ABL fusion oncoprotein expressed in chronic myelogenous leukemia (CML). It also cross-reacts with other tyrosine kinases, such as c-ABL, c-KIT, and platelet-derived growth factor receptors (PDGFRs), and has been used for the treatment of gastrointestinal stromal tumor, small cell lung cancer, ovarian cancer, and colorectal carcinoma (9 –13). In preclinical cancer models, we have already reported the partial effectiveness of imatinib on ATC cells (14). We have also demonstrated that imatinib combined with ionizing radiation enhanced senescence-like growth arrest (SLGA) (15). Very recently, a clinical trial using imatinib as a single agent for the treatment of ATC has shown weak responses: no complete response, 25% partial response, and 50% stable disease at 8 weeks, but the rate of 6-month survival was only 45% (16).
Several reports have demonstrated that imatinib enhanced the sensitivity of various cancer cells to anticancer drugs (9,17). A few studies have investigated the anticancer efficacy of imatinib/docetaxel combinations in preclinical models. It was shown that the combination of imatinib and docetaxel was significantly more effective than either agent alone in the nonsmall cell lung carcinoma xenograft model. In this model, imatinib, as an inhibitor of PDGFRβ, decreased microvessel density and interstitial fluid pressure, and thereby improved subsequent delivery of docetaxel (18). Another report showed that in human CML cells, the imatinib/docetaxel combination induced apoptosis through decreasing mitochondrial membrane potential and increasing caspase-3 enzyme activity (19). Kinsella et al. reported that imatinib combined with docetaxel strongly inhibited both proliferation and invasion, and had a proapoptotic effect in glioma cells (20). So far, there has been no report regarding the therapeutic efficacy of imatinib/docetaxel combinations in ATC.
In this study, we demonstrate that imatinib enhanced the antitumor activity of docetaxel in ATC cells. Docetaxel seemed to induce both proapoptotic and antiapoptotic signaling pathways, and imatinib blocked the antiapoptotic signal through inhibition of docetaxel-induced nuclear factor-κB (NF-κB) activation.
Methods
Reagents
Imatinib (Novartis, Basel, Switzerland) was dissolved in dimethyl sulfoxide (DMSO) at stock concentration of 7 mM for in vitro experiments, and for in vivo experiments, imatinib tablets were dissolved in distilled water, and insoluble material was removed by repeated centrifugation at 2500 g as described previously (21). Docetaxel (Wako Chemicals, Osaka, Japan) was dissolved in DMSO at a stock concentration of 1 mM. The antibodies to p65, survivin, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); the antibodies to X-linked inhibitor of apoptosis (XIAP), cleaved caspase-3, poly (ADP-ribose) polymerase (PARP), IκBα [nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha], and antirabbit and antimouse horseradish peroxidase–conjugated antibodies were from Cell Signaling Technology (Beverly, MA).
Cell culture
The human ATC cell line FRO was originally provided by Dr. James A. Fagin (University of Cincinnati College of Medicine, Cincinnati, OH; currently Memorial Sloan-Kettering Cancer Center, New York, NY), and KTC-2 was from Dr. Kurebayashi (Kawasaki Medical School, Kurashiki, Japan) (22). All cells were grown in RPMI 1640 (Wako Chemicals) supplemented with 5% (v/v) fetal bovine serum and 1% (w/v) penicillin/streptomycin (Wako Chemicals).
Cell growth assay
Cells were seeded onto each well of 24-well plate (500 μL, 15×103 cells per well) and incubated for 24 hours before treatment. Solutions containing various concentrations of docetaxel and/or imatinib were added to each well in 55 μL medium, with three wells used for each concentration. In the control wells, DMSO was added. The final concentration of DMSO in any well did not exceed 0.2% (v/v). After incubation, the number of cells were counted with a Coulter counter (Beckman Coulter, Fuller, CA).
Flow cytometry analysis with the annexin V/PI staining
Detection of apoptotic cells was performed with an Annexin V-PI apoptosis detection kit (Wako Chemicals) according to the manufacturer's instructions. In brief, 4×105 cells were double stained with fluorescein isothiocyanate–conjugated annexin V and propidium iodide (PI) for 15 min at room temperature in a Ca2+-enriched binding buffer and then analyzed on an FACS Vantage SE (BD Biosciences, San Jose, CA). Fluorescein isothiocyanate and PI emissions were detected in the FL-1 and FL-3 channels, respectively. Analysis was done with Cell Quest software (BD Biosciences).
Senescence-associated β-galactosidase staining
Senescence-associated β-gal staining (SA-β-gal) was performed as described elsewhere (23). Briefly, after experimental treatment, cells on plates were fixed with 2% (v/v) formaldehyde/0.2% (v/v) glutaraldehyde, washed with phosphate-buffered saline, and assayed for SA-β-gal activity using X-gal (5-bromo-4-chloro-3-indolyl β-D-galactosidase) at pH 6.0. SA-β-gal+ cells were detected by bright-field microscopy.
Western blotting
Forty micrograms of protein was separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA). After incubation with an appropriate primary antibody, the antigen–antibody complexes were visualized using horseradish peroxidase–conjugated secondary antibody and a chemiluminescence system (NacalaiTesque, Kyoto, Japan). Detection was performed using an LAS3000 imaging system (FUJIFILM, Tokyo, Japan).
DNA-binding assay
The multiwell colorimetric assay for active NF-κB was performed as described previously (24). Briefly, equal amount of nuclear extracts was incubated in 96-well plate coated with immobilized oligonucleotide containing a NF-κB consensus binding site. NF-κB binding to the target oligonucleotide was detected with primary antibody specific for p65 subunit and horseradish peroxidase–conjugated secondary antibody. For quantification of activity, optical densities (ODs) were measured at 450 nm using a microplate reader 2030 ARVO X (Perkin Elmer, Inc., Waltham, MA).
In vivo xenograft model
All mice were maintained at Nagasaki University animal facility, and all animal experiments described in this study were conducted in accordance with the principles and procedures outlined in the Guide for the Care and Use of Laboratory Animals of Nagasaki University. FRO cells (3×106) resuspended in RPMI 1640 were injected subcutaneously into both flanks of 6-week-old male BALB/c nu/nu mice (CLEA Japan, Tokyo, Japan), five animals per group. Then, they were randomly assigned into four groups. The tumor sizes were measured every 3 days with calipers, and tumor volumes were calculated according to the formula, a 2×b×0.4, where a is the smallest tumor diameter and b is the diameter perpendicular to a. Imatinib solution in sterile water/phosphate-buffered saline (ratio 1:1) was injected intraperitoneally (i.p.) daily for 2 weeks at a dose of 50 mg/kg beginning from day 9 after tumor implantation. Docetaxel, diluted in phosphate-buffered saline/DMSO (ratio 1:1), was injected i.p. at a dose of 5 mg/kg on days 9 and 16. Combined treatment mice were given both drugs. Control group mice received vehicle injections only. For 27 days, tumor size was monitored, and body weight, feeding behavior, and motor activity of each animal were monitored as indicators of general health.
Statistical analysis
All data were expressed as the mean±standard deviation. Differences between groups were examined for statistical significance with analysis of variance followed by Tukey's posttest. A p-value not exceeding 0.05 was considered statistically significant.
Results
Inhibition of cell growth by docetaxel and/or imatinib
To investigate the effect of docetaxel and/or imatinib, cell growth assays were done. Our previous experiments (7,14) (unpublished data, Michiko Matsuse, Nagasaki University, 2008) demonstrated that moderate cell growth inhibition by imatinib and docetaxel was observed at concentrations of 7 μM and 1–2 nM in FRO cells, respectively. Thus, we conducted the experiments using 7 μM of imatinib and 1 nM of docetaxel. As shown in Fig. 1, both docetaxel and imatinib had a moderate inhibitory effect on the growth of FRO cells, and combined treatment significantly reduced the cell number compared with single treatment (Fig. 1). We also used another ATC cell line, KTC-2 cells, in which the growth inhibition by imatinib was less effective compared with FRO cells, and the potency of docetaxel was more pronounced. The combined treatment similarly inhibited cell growth, almost completely (Fig. 1). The rates of growth reduction at 4 days in docetaxel, imatinib, and combined treatment were 47.3%, 68.5%, and 96.0% in FRO cells and 76.1%, 29.6%, and 99.5% in KTC-2 cells, respectively (Fig. 1).

Cytotoxic effect of docetaxel and/or imatinib on anaplastic thyroid cancer (ATC) cells. Growth of FRO and KTC-2 cells treated with 1 nM of docetaxel and/or 7 μM of imatinib for 2 and 4 days was determined by cell count assay. Each point represents mean±standard deviation. *p<0.01 versus any other group. Similar results were obtained in three independent experiments.
Apoptotic changes in cells treated with the drugs
Although the degree of cell detachment was more notable in the combined treatment than in the single treatment group, we first explored the possibility of involvement of senescence-like growth arrest (SLGA) because we previously demonstrated that imatinib plus radiation therapy enhanced SLGA in ATC cells (15). Neither single treatment nor combined treatment induced SA-β-gal activity in FRO or KTC-2 cell lines, suggesting that the growth inhibitory effect was not due to SLGA (data not shown). Next, we examined whether the effect was associated with apoptosis. Cells were treated with the drugs for 16 hours and then double stained with fluorescein isothiocyanate–conjugated annexin V and PI to look at early apoptotic response (right lower quadrant in each dot plot in Fig. 2) and subsequent cell death (right upper quadrant). Imatinib barely caused apoptosis, and docetaxel moderately induced it. On the other hand, combined treatment further increased apoptosis (Fig. 2).

Apoptotic changes in cells treated with drugs. FRO cells were treated with the drugs (1 nM docetaxel and/or 7 μM imatinib) for 16 hours and then double stained with fluorescein isothiocyanate–conjugated annexin V and propidium iodide (PI), then analyzed on an FACS Vantage SE. Fluorescein isothiocyanate and PI emissions were detected in the FL-1 and FL-3 channels, respectively. The cells in left lower, right lower, and right upper quadrant represent viable cells, early apoptotic cells, and terminal stage of apoptotic or necrotic cells, respectively. Data are representative of two independent experiments.
Effects of docetaxel and imatinib on proapoptotic and antiapoptotic factors
We next examined the status of several key proteins involved in apoptosis by Western blotting. After ATC cells were treated with docetaxel, the cleavages of caspase-3 (19 kDa and 17 kDa) and PARP (89 kDa) levels were induced (Fig. 3). The cleaved PARP and caspase-3 were further increased by combined treatment, suggesting stronger apoptotic change (Fig. 3). XIAP and survivin belong to the human inhibitors of apoptosis (IAP) family, and their overexpression in cancer cells suggests an important role for these proteins in cancer progression. We tested whether docetaxel and/or imatinib modulate the expression of these antiapoptotic gene products by Western blotting. As shown in Fig. 3, although ATC cells themselves expressed high levels of XIAP and survivin, docetaxel further increased the levels (Fig. 3). A single treatment of imatinib did not change their background levels. However, the increased docetaxel-induced XIAP and survivin expression was suppressed by the combination treatment (Fig. 3).
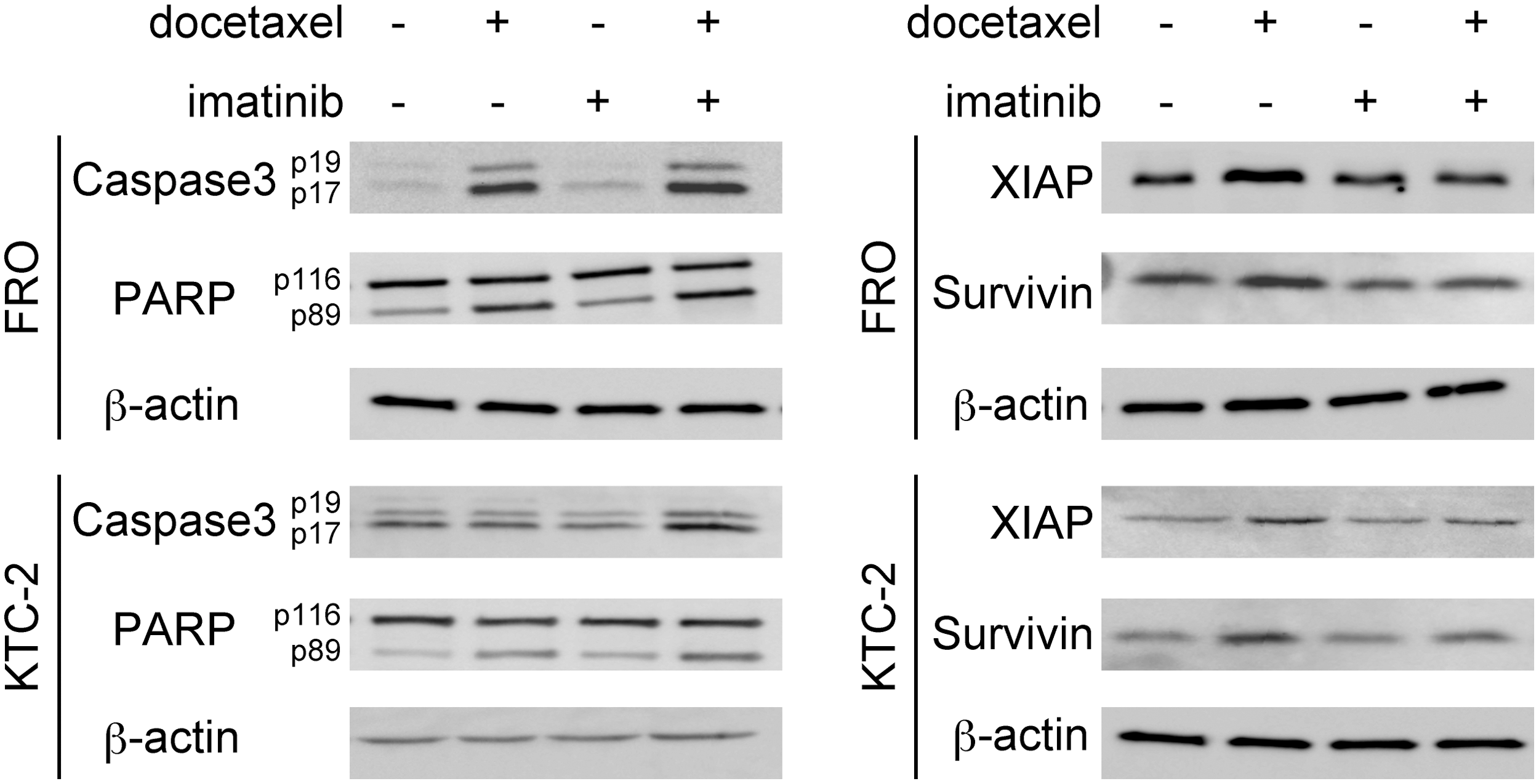
Effects of docetaxel and imatinib on apoptotic factors. Cells were treated with the drugs (4 nM docetaxel and/or 7 μM imatinib) for 16 hours [X-linked inhibitor of apoptosis (XIAP) and survivin] or 24 hours [caspase 3 and poly (ADP-ribose) polymerase (PARP)], and whole-cell lysates were examined by Western blotting. β-Actin was used as a loading control. Data are representative of at least two independent experiments.
Docetaxel induces NF-κB activation, and imatinib inhibits the effect
Since we already reported that the expression of those antiapoptotic factors was regulated through NF-κB signaling pathway in ATC cells (7), we next performed DNA-binding assays using nuclear extracts to assess NF-κB activity. In both FRO and KTC-2 cell lines, the binding activity of nuclear p65 was increased by docetaxel treatment. Although imatinib alone had no effect on NF-κB background levels, with the combined treatment, imatinib significantly suppressed docetaxel-induced NF-κB activation (Fig. 4A). A similar trend was observed in the amount of nuclear p65 (Fig. 4B). We also checked IκBα expression. As shown in Fig. 4B, combined treatment reduced IκBα protein level, consistent with our previous studies (7,25). NF-κB is known to bind the IκBα promoter and activate its synthesis, and therefore the inhibition of NF-κB probably suppressed de novo synthesis of IκBα. Presumably, for the same reason, IκBα expression after docetaxel treatment was not changed (Fig. 4B).
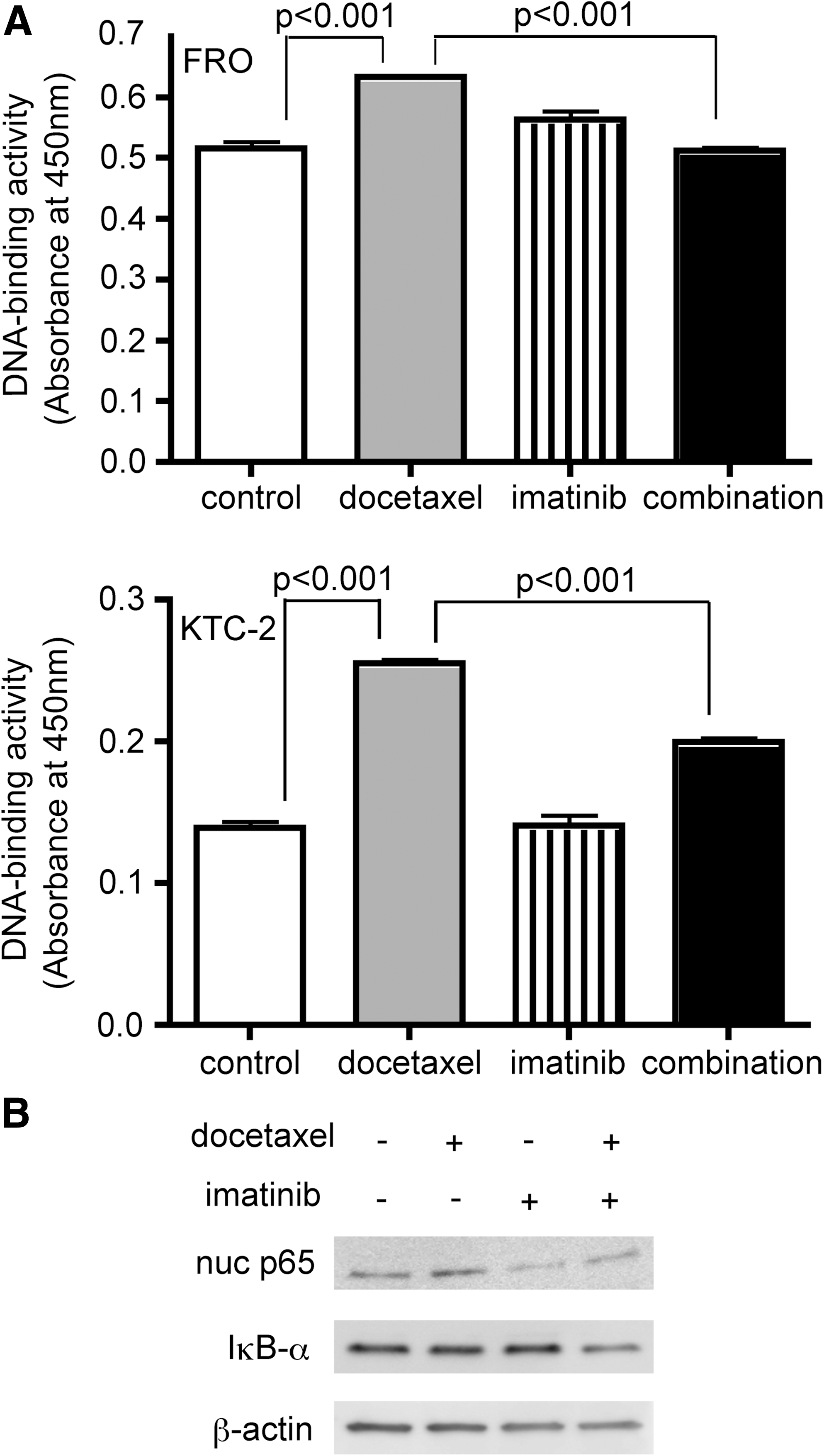
Docetaxel induces nuclear factor-κB (NF-κB) activation, and imatinib inhibits the effect.
In vivo effects of the combined treatment with docetaxel and imatinib
To explore the effects of the combined treatment in vivo, we used an animal xenograft model inoculated with FRO cells, and the treatments were done as described in the Materials and Methods section. As shown in Fig. 5, the mean tumor size of the imatinib-treated group was smaller than that of control, but this difference was not statistically significant. Although docetaxel treatment alone was able to delay tumor growth moderately, the effect of the combined treatment with docetaxel and imatinib was far greater. Body weight and physical activity of mice exposed to these drugs were not significantly affected.
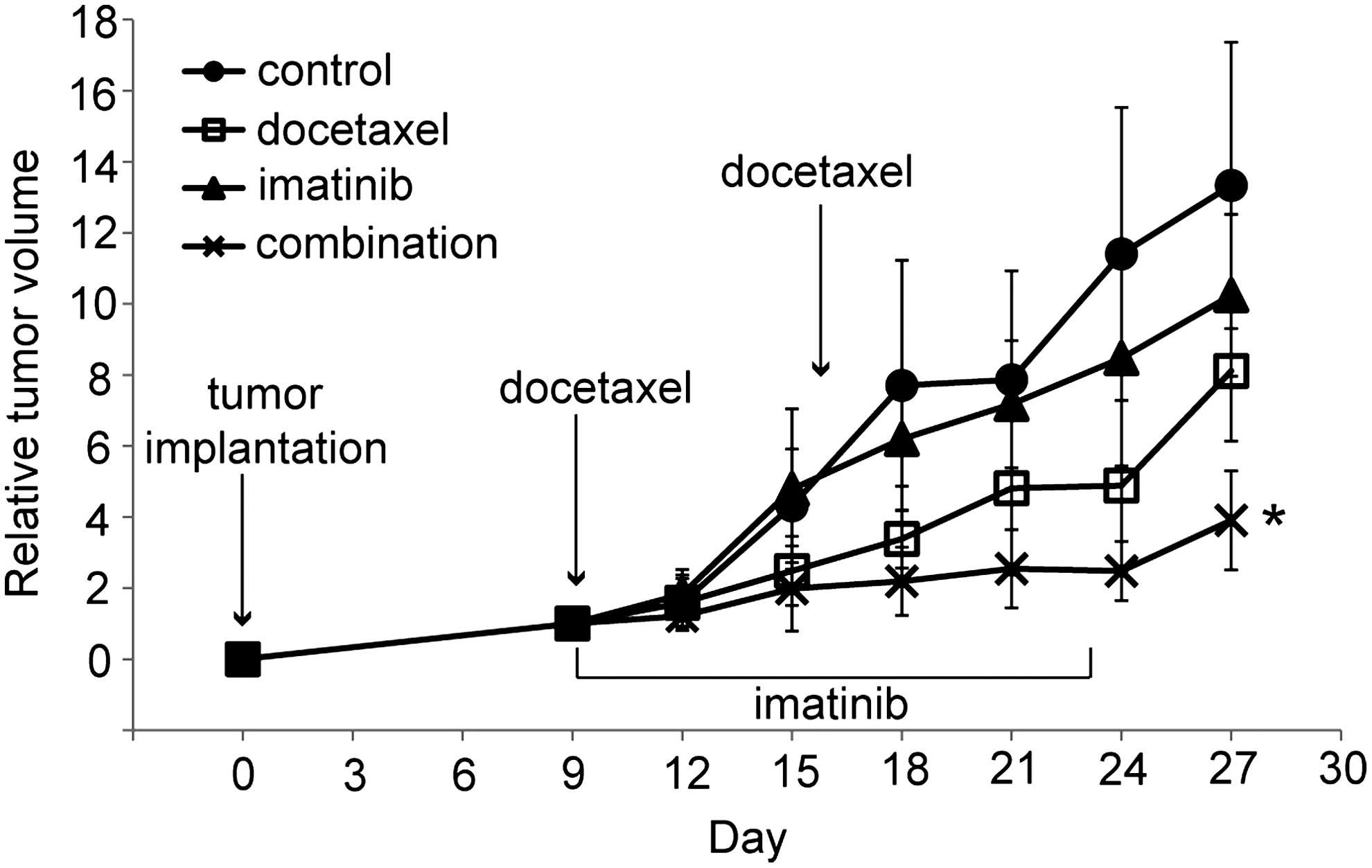
Effect of docetaxel and imatinib in FRO tumor xenograft model. FRO cells were implanted and imatinib was injected i.p. at a dose of 50 mg/kg/day for 14 days, beginning on day 9 after tumor implantation. Docetaxel was injected i.p. at a dose of 5 mg/kg on days 9 and 16. Combined treatment mice were given both drugs. Control group mice received vehicle injection only. Each point represents mean±SD of 10 tumors (in five mice). *p<0.01 versus any other group. i.p., intraperitoneally.
Discussion
We and other groups have reported that taxanes (including paclitaxel and docetaxel) induce both pro- and antiapoptotic signaling pathways, and that antiapoptotic factors are also induced, through NF-κB activation (7,26,27). NF-κB is a transcription factor that regulates genes involved in cellular proliferation and survival (28). In most cases, NF-κB suppresses apoptosis by upregulation of antiapoptotic proteins, including IAP, such as c-IAP, B-cell lymphoma-extra large (BCL-xL), XIAP, and survivin (29,30). In addition, basal NF-κB activity is often increased in various types of human cancers, which causes chemotherapy resistance (31). Under certain conditions, cytotoxic drugs, such as taxanes, induce NF-κB activation in different types of malignant cells (26,32 –36). The present data also showed that docetaxel induced NF-κB activation and then upregulated antiapoptotic factors in two ATC cell lines, FRO and KTC-2.
In our experiments, imatinib seemed to enhance apoptosis presumably through the inhibition of docetaxel-induced NF-κB activation. There are several reports proposing the mechanism by which imatinib inhibits the docetaxel-induced NF-κB activation. The phosphoinositide 3-kinase (PI3K)/AKT (also known as Protein Kinase B [PKB]) pathway has been associated with cancer cell resistance to chemotherapeutic drugs including paclitaxel (34,37). Possible mechanisms of such resistance may be due to the activation of the NF-κB pathway via PI3K/AKT activation (38). Qian et al. compared the gene expression profiles in individual human prostate cancer specimens before and after chemotherapy and showed that docetaxel treatment increased CCL2 expression (39). They also showed that upregulation of CCL2 contributed to chemotherapy resistance through stimulating mitogen-activated protein kinase and PI3K/AKT signaling pathways. Several other reports have demonstrated that imatinib inhibited the PI3K/AKT pathway, resulting in the inhibition of NF-κB activation in cancer cells. Fang et al. reported that imatinib induced apoptosis in BCR/ABL-positive human leukemia cells in association with the downregulation of antiapoptotic factors, such as XIAP, through the inhibition of AKT and NF-κB activities (40). Xu et al. reported that imatinib inhibited ionizing radiation–induced RelB nuclear translocation by decreasing the phosphorylation levels of PI3K (Tyr458) and AKT (Ser473) in androgen-independent prostate cancer cells (41). They showed that imatinib inhibited PI3K tyrosine phosphorylation, leading to the downregulation of the AKT/IKK-α–activated NF-κB pathway. We examined whether docetaxel and/or imatinib modulate the phosphorylation level of AKT. However, we could detect neither the activation of AKT by docetaxel treatment nor the inhibition of AKT by imatinib (data not shown). Further experiments are necessary to clarify the exact mechanism by which imatinib inhibits the docetaxel-induced NF-κB activation in ATC cells.
Data obtained in our experiments showed that combination of docetaxel and imatinib effectively killed ATC cells, both in vitro and in vivo. Based on our and other's findings, we propose a mechanistic scheme in Fig. 6. As previously shown, docetaxel activates both proapoptotic and antiapoptotic signals. In proapoptotic pathway, docetaxel binds to microtubules, impairs mitosis, and induces apoptosis. In antiapoptotic pathway, docetaxel also induces NF-κB activation and in turn increases the expression of antiapoptotic molecules. Imatinib inhibits docetaxel-induced NF-κB activation (but does not reduce basal NF-κB levels, even though they are high). By this mechanism, imatinib presumably modulates the balance between pro- and antiapoptotic signals and enhances docetaxel-induced apoptosis.
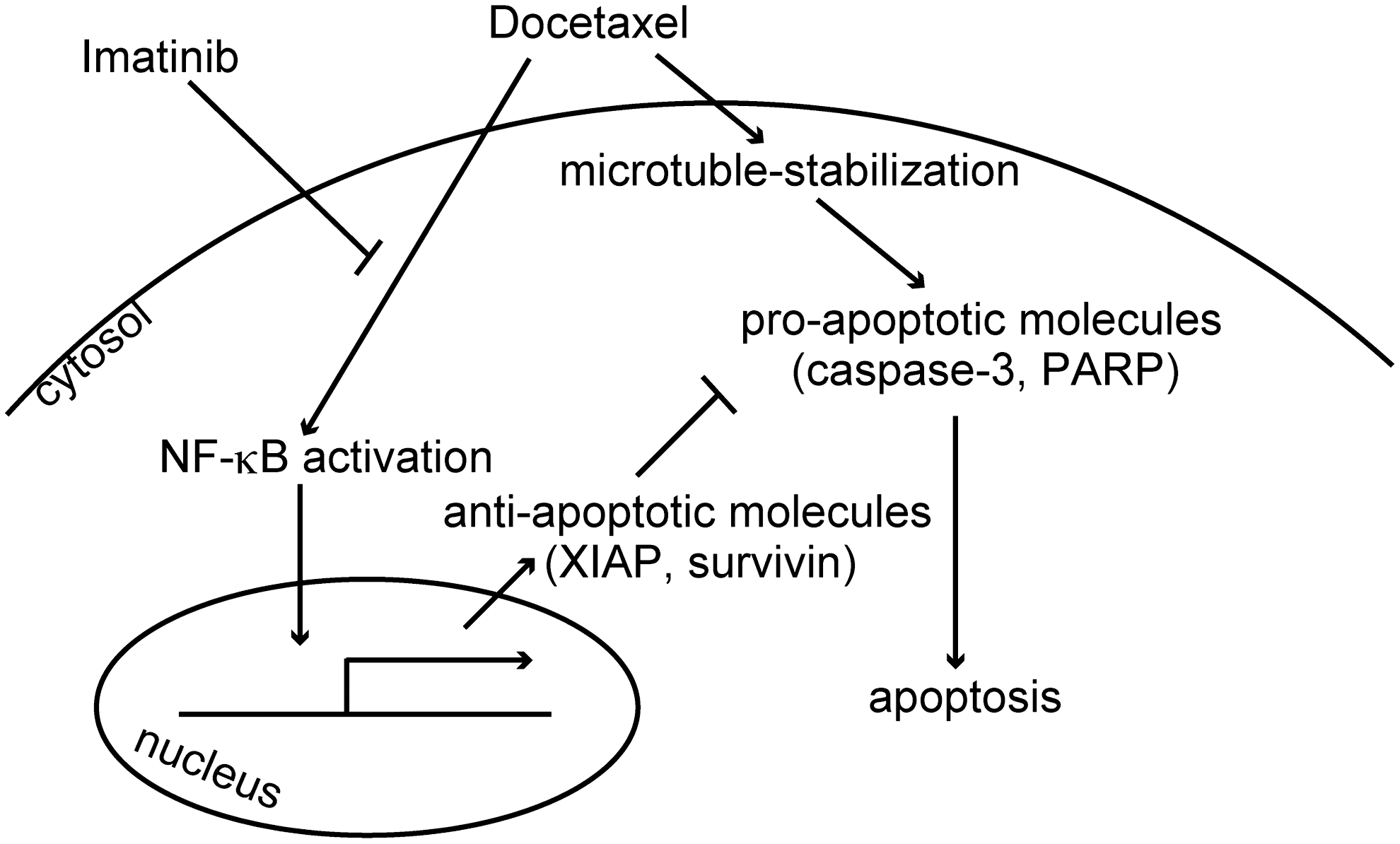
A proposed mechanistic model of enhancement of docetaxel-induced apoptosis by imatinib.
In conclusion, our present study demonstrates that imatinib enhanced antitumor activity of docetaxel in ATC cells, suggesting that this combination may be a promising approach for the treatment of ATCs. Since docetaxel and imatinib have already been approved and currently being used for other type of cancers, this combination strategy can be rapidly applied in clinical trials.
Footnotes
Acknowledgments
This work was supported in part by Grant-in-Aid for Scientific Research (#23591357, #22256004, and #22390189), Grant-in-Aid for Young Scientists (#22791204), and Global COE Program from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Disclosure Statement
The authors declare no conflict of interest.