
Abstract
Background:
In the thyroid, iodine deficiency (ID) induces angiogenesis via a tightly controlled reactive oxygen species (ROS)–hypoxia inducible factor-1 (HIF-1)–vascular endothelial growth factor (VEGF) dependent pathway (ROS-HIF-VEGF). Deficient iodine intake may be associated with increased thyroid cancer incidence. The hypothesis of this work is to test whether ID affects the angiogenic processes in thyroid malignant cells by altering the ROS-HIF-VEGF pathway.
Methods:
Goiters were obtained in RET/PTC3 transgenic and wild-type (wt) mice and ID was induced in three thyroid carcinoma cell lines (TPC-1, 8305c, and R082-w1). Thyroid blood flow, VEGF mRNA and protein, and HIF-1α protein expression were measured. The role of HIF-1 and of ROS was assessed using echinomycin and N-acetylcysteine (NAC), respectively.
Results:
The goitrogen treatment increased the thyroid blood flow in wt and RET/PTC3 mice. Compared with wt mice, basal VEGF expression was higher in RET/PTC3 mice and increased with goitrogen treatment. In the three cell lines, ID induced marked increases in VEGF mRNA, and moderate increases in HIF-1α protein expression that were not transient as in normal cells. ID-induced VEGF mRNA expression was fully (8305c), partially (TPC-1), or not (R082-w1) blocked by echinomycin. NAC had no effect on ID-induced VEGF mRNA and HIF-1α protein expression in the three cell lines.
Conclusions:
ID induces a long lasting angiogenic phenotype in thyroid cancer cells that occurs through VEGF induction via a pathway partially mediated by HIF-1, but not by ROS. These results suggest that, in contrast with normal cells, ID-induced angiogenesis in cancer cells occurs via alternative and likely less controlled routes, thereby leading to uncontrolled growth.
Introduction
The incorporation of iodine into thyroglobulin is required for thyroid hormone synthesis, a process activated by TSH. But TSH intervenes rather late in the course of ID, only when intrathyroidal iodide stores are insufficient to fulfill thyroid hormone synthesis. Thus, thyrocytes use various TSH-independent autoregulatory mechanisms to adapt to daily variations in iodide supply. These mechanisms include the stimulation of iodide trapping by sodium-iodide symporter (NIS), the recycling of intracellular iodine by iodotyrosine dehalogenase 1 (DEHAL1), the preferential synthesis of T3 over T4, and the peripheral conversion of T4 into T3 (10 –12). An alternative mechanism by which thyrocytes adapt to iodine fluctuations is the regulation of their own vascular network. Each follicle forms with its adjacent microvasculature a highly sophisticated structure, the angio-follicular unit that is now recognized as the true functional and morphological unit of the thyroid (13 –17). By enhancing the local clearance of iodine, changes in the microcirculation contribute to counteract ID. We previously showed that in case of ID, a nearly instantaneous TSH-independent angiogenic reaction occurs in the thyroid. It lasts at least a week before a second phase of more robust angiogenesis under TSH control (18). This highly controlled process leads to a harmonious and well-controlled synchronization between epithelial and endothelial compartments to safeguard thyroid hormone synthesis.
As lately shown, the angiogenic signal, identified as vascular endothelial growth factor (VEGF), is initiated within the epithelial cells via a pathway mediated by reactive oxygen species (ROS) and the hypoxia inducible factor-1 (HIF-1) (19). Of note, this ID-induced pathway is activated to optimize the functional performance of thyrocytes. It is transient and is not associated with abnormal growth of tissues, which is under tight control (18,19).
Because anarchic angiogenesis is a hallmark of carcinogenesis, we hypothesized that ID promotes the development of thyroid cancers by altering the ROS-HIF-VEGF pathway. To test this hypothesis, an in vivo model of RET/PTC3 mice and three in vitro models of thyroid cancer cells were used to isolate ID as the sole factor that could be accountable if the angiogenic process was noted to be altered.
Materials and Methods
Animals and treatments
ID was induced in 6-month-old transgenic RET/PTC mice, bearing the RET/PTC3 translocation under the control of bovine thyroglobulin promoter (developed by Massimo Santoro's group at DBPCM [Department of Cellular and Molecular Biology and Pathology], Naples, Italy, and kindly provided by Jacques Dumont, ULB, Brussels, Belgium), and wild-type (wt) C57BL/6 mice by feeding a low-iodine diet (LID<20 μg iodine/kg; Animalabo) supplemented with sodium perchlorate 1% in drinking water for 2 days. Control animals received normal diet and tap water. Mice were housed and handled according to the Belgian regulation of Laboratory Animal Welfare.
Laser Doppler blood flow measurement and preparation of tissue samples
Thyroid blood flow was measured using a Laser Doppler imager (Moor Instruments) in anesthetized mice as previously described (20). Thyroid lobes were identified and delineated based on anatomical images and blood flow was quantified in the areas of interest from colored histogram pixels (18). After measurements, both thyroid lobes were removed. One was fixed in paraformaldehyde (4% in phosphate-buffered saline [PBS]) for 36 hours and embedded in paraffin. Thick sections (5 μm) were used for immunohistochemistry. The second lobe was directly frozen in liquid nitrogen and used for RNA analysis.
Cell cultures
Three human thyroid carcinoma cell lines were used. They were chosen because they were shown to keep characteristics of cells derived from thyroid cancers with associated mutations and expression of Pax8 or Titf1 (21). TPC-1 (thyroid papillary carcinoma with a RET/PTC1 translocation and expressing Pax8, but not Titf1) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS); 8305C (undifferentiated thyroid carcinoma, with a p53 mutation, expressing Titf1, but not Pax8) cells were grown in MEM supplemented with 10% FBS, 2 mM L-glutamine, and 1% nonessential amino acid; and R082-w-1 (thyroid follicular carcinoma, with a BRAF mutation, expressing Pax8, but not Titf1) cells were grown in DMEM+Ham's F12+MCDB 105 (2:1:1) medium supplemented with 10% FBS and 2 mM L-glutamine. The three cell lines were compared with PCCL3 cells (normal rat thyroid cells, a gift from Dr. F. Miot, IRIBHN). As previous results (19) using PCCL3 cells and human thyrocytes in primary cultures showed that cellular changes in response to ID were strictly identical in both models, we decided to use only PCCL3 cells for comparison. PCCL3 cells were grown in Coon's modified Ham's F12 medium supplemented with 5% newborn calf serum, 5 mU/mL TSH, 10 μg/mL insulin, and 5 μg/mL transferrin. All cells were grown to 80%–90% confluence in media containing NaI at a concentration of 10−8 M in a humidified atmosphere (5% CO2). ID was induced as previously described (19). Briefly, culture media were replaced the day of the experiment by fresh media with NaI (control), or without NaI. Biological samples were then harvested after 2, 4, or 6 hours corresponding to the experimental times. To study the role played by HIF-1α and ROS, cells were treated either with echinomycin (100 nM; Alexis Biochemicals), to inhibit HIF-1 binding to the hypoxia response element (HRE) located on the VEGF gene (22), or with N-acetylcysteine (NAC, 1 mM; Sigma), a potent antioxidant.
Quantitative polymerase chain reaction
For in vitro samples, cells from five individual wells for each condition were collected in TriPure isolation reagent (Roche Diagnostics GmbH). For in vivo samples, each frozen thyroid lobe was homogenized in TriPure isolation reagent. Total RNA purification and reverse transcription were performed as previously described (23). cDNAs (2 μL) were mixed with 500 nM of each selected primer (see Table 1) and SYBR Green reaction mix (Quanta BioScience) in a final volume of 25 μL. Reactions were performed in an iCycler (IQ5; Bio-Rad) using the following program: 95°C/1.5 min, followed by 40 cycles of 95°C/15 s, annealing temperature (see Table 1)/45 s, and 81°C/15 s. Amplification levels were normalized to those of β-actin.
VEGF, vascular endothelial growth factor; TPO, thyroid peroxidase; NIS, sodium-iodide symporter.
HIF-1α western blotting
Thyrocytes were suspended in 120 μL of lysis buffer (50 mM imidazole, 300 mM KCl, 10 mM NaF, 1 mM EDTA, 0.5 mM MgCl2, 10 mM D-glycerophosphate, 1 mM Na3VO4, 1 mM DTT, 0.1 mM PMSF, and 1 mM benzamidine [pH 7]), and immediately frozen in liquid nitrogen. Forty microliters of 4× Laemmli buffer was then added. The loading buffer (10 μL of Laemmli containing 100 mM DTT and 0.1% bromophenol blue) was added to the protein solution. Proteins were separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Membranes were blocked for 1 hour at room temperature (RT) in PBS, 5% nonfat dry milk, and 0.1% Tween and incubated overnight at 4°C with a polyclonal antibody raised against HIF-1α (BD Biosciences) at a dilution of 1/250. Membranes were washed with PBS-Tween 0.1%, incubated for 1 hour at RT with peroxidase-labeled secondary antibody (Pierce), and visualized with enhanced chemiluminescence (SuperSignal West Femto, Pierce) on CL-Xposure films (Pierce). The same membranes were also treated with an anti-β-actin antibody (1/8000; Sigma).
Immunohistochemistry
Tissue sections were washed with PBS supplemented with 1% bovine serum albumin (PBS-BSA) and incubated at RT for 30 minutes with normal goat serum (1/50; Vector Laboratories, Burlingame, CA) in PBS-BSA. Slides were then incubated at RT with primary antibodies (anti-VEGF antibody, 1/50; Santa Cruz Biotechnology, Santa Cruz, CA, or anti-highly iodinated thyroglobulin, 1/4000; a gift from JJM de Vijlder, Amsterdam). The first antibody was detected using an Envision mouse secondary antibody (DakoCytomation), or an ABC Perox Kit (Vector Laboratories). The peroxidase activity was revealed using AEC (3-amino-9-ethylcarbazole; DakoCytomation) as substrate. Sections were counterstained with Mayer's hematoxylin. For VEGF detection, tissue sections were pretreated in a microwave oven in citrate buffer (0.01 M, pH 6) for one cycle of 3 minutes at 750 W, followed by 4 cycles of 3.5 minutes each at 350 W. Negative controls were performed by omitting the primary antibody and by adding a neutralizing peptide in excess for VEGF (SantaCruz), or T4 for highly iodinated thyroglobulin.
Data analysis and statistics
All data are expressed as mean±SEM (n=6 for in vivo experiments and n=5 for in vitro experiments). Western blots were scanned and quantified by densitometry using the NIH Scion Image Analysis Software (National Institutes of Health). Western blots and quantitative polymerase chain reaction data were normalized to β-actin. Statistical analyses were performed using analysis of variance followed by a Tukey-Kramer or Newman-Keuls multiple comparison post hoc tests where convenient, and the ID groups were compared with controls using unpaired t-test (GraphPad Instat). p<0.05 was considered statistically significant.
Results
ID increases the thyroid blood flow and VEGF expression in RET/PTC3 mice
We first tested the effects of a short-term goitrogen treatment on the expression of thyroid-specific proteins. In wt mice, as already reported (18), a 2-day goitrogen treatment affected neither thyroid peroxidase (TPO) mRNA expression, nor the expression of highly iodinated thyroglobulin, a marker of thyroid hormone synthesis. The same results were obtained in RET/PTC3 mice (data not shown). These results confirm that after 2 days of a goitrogen treatment, the thyroid gland of wt or RET/PTC3 mice was not stimulated by TSH; any change observed using this model being therefore attributable only to ID.
To test whether ID promotes angiogenesis in thyroid cancers, we first measured the thyroid blood flow in RET/PTC3 mice as compared with wt mice. In wt mice, the thyroid blood flow was increased after 2 days of goitrogen treatment. In RET/PTC3 mice, the basal thyroid blood flow was similar to that in wt mice. In RET/PTC3 mice, goitrogen treatment induced a significant increase in the thyroid blood flow that was similar to that in wt mice (Fig. 1A).
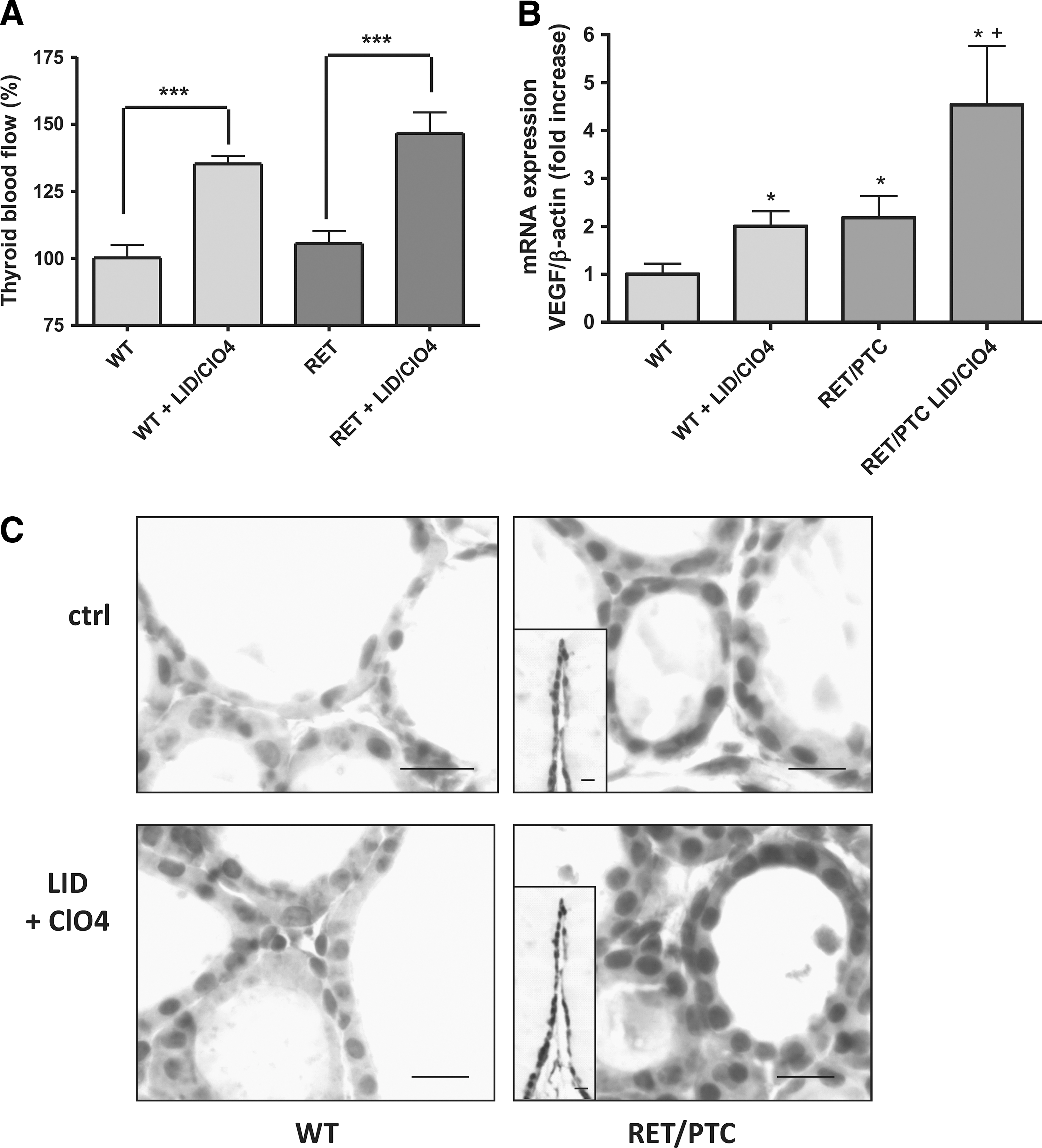
Iodine deficiency (ID)–induced increase in the thyroid blood flow and in vascular endothelial growth factor (VEGF) mRNA and protein expression in wild-type (wt) C57BL and RET/PTC3 mice. C57BL (wt) and RET/PTC3 (RET) mice received a goitrogen treatment (low-iodine diet [LID]+ClO4) for 2 days.
Basal levels of VEGF mRNA and protein expression were higher in RET/PTC3 mice than in wt mice. VEGF mRNA expression was increased by the goitrogen treatment in both mouse types (Fig. 1B). VEGF protein expression analyzed by immunohistochemistry was detected at low level in thyrocytes of wt mice, and this expression was increased by the goitrogen treatment. In RET/PTC3 mice, protein expression was more heterogeneous, some cells being highly positive while others only displaying faint staining. In addition, staining was observed in the surrounding connective tissue. The staining was somewhat reinforced in thyrocytes by the goitrogen treatment (Fig. 1C). We also checked for the expression of VEGF receptor mRNAs, Flk1/VEGFR1 and Flt1/VEGFR2. Both receptors were expressed at the same level in treated and nontreated wt mice. Nevertheless, while Flk1/VEGFR1 displayed the same level of expression in RET/PTC3 mice, without any change after 2 days of ID, Flt1/VEGFR2 mRNA expression was lower in RET/PTC3 mice than in wt mice (Fig. 2). After 2 days of ID, this expression was not significantly increased (Fig. 2).
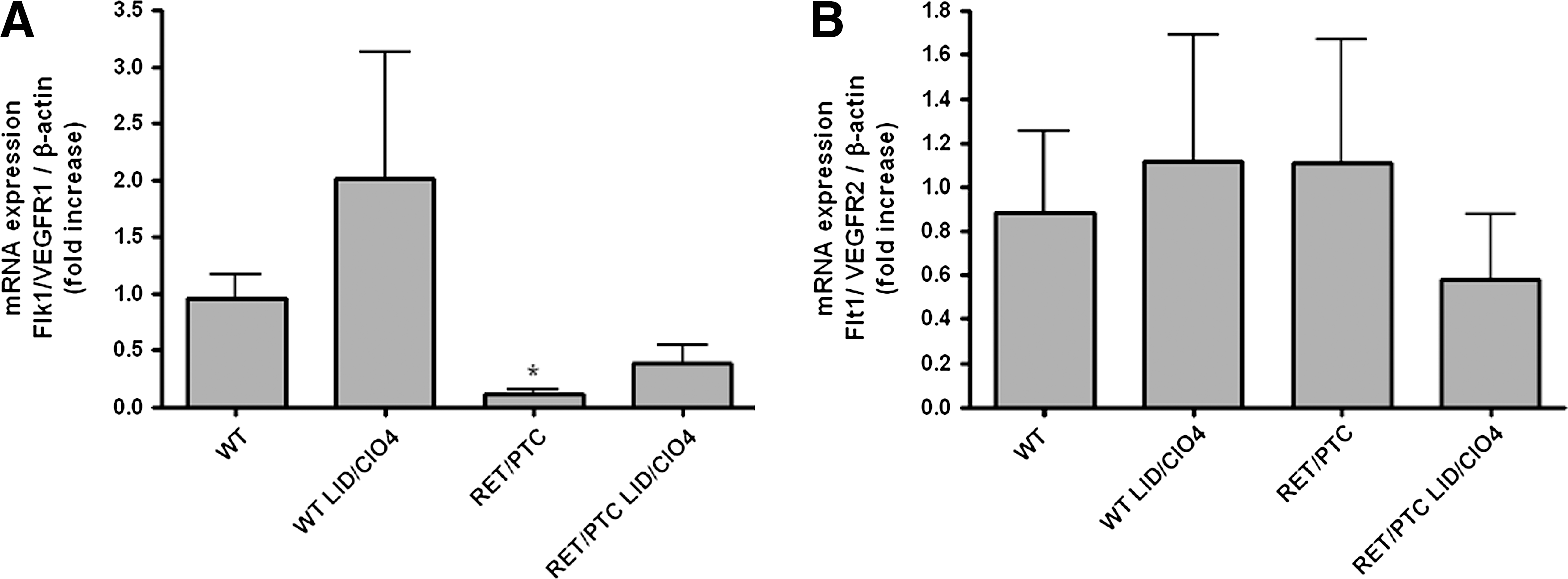
ID-induced changes in VEGF receptor mRNA expression in wt and RET/PTC3 mice. C57BL wt and RET/PTC3 (RET) mice received a goitrogen treatment (LID+ClO4) for 2 days. Flk1/VEGFR1
ID increases NIS mRNA expression both in vivo and in vitro
In wt mice, the 2-day goitrogen treatment induced a significant increase in NIS mRNA expression. RET/PTC3 mice expressed the same basal level of NIS mRNA, but the 2-day goitrogen treatment did not induce a significant increase in NIS mRNA level even though a positive trend was observed (Fig. 3A).
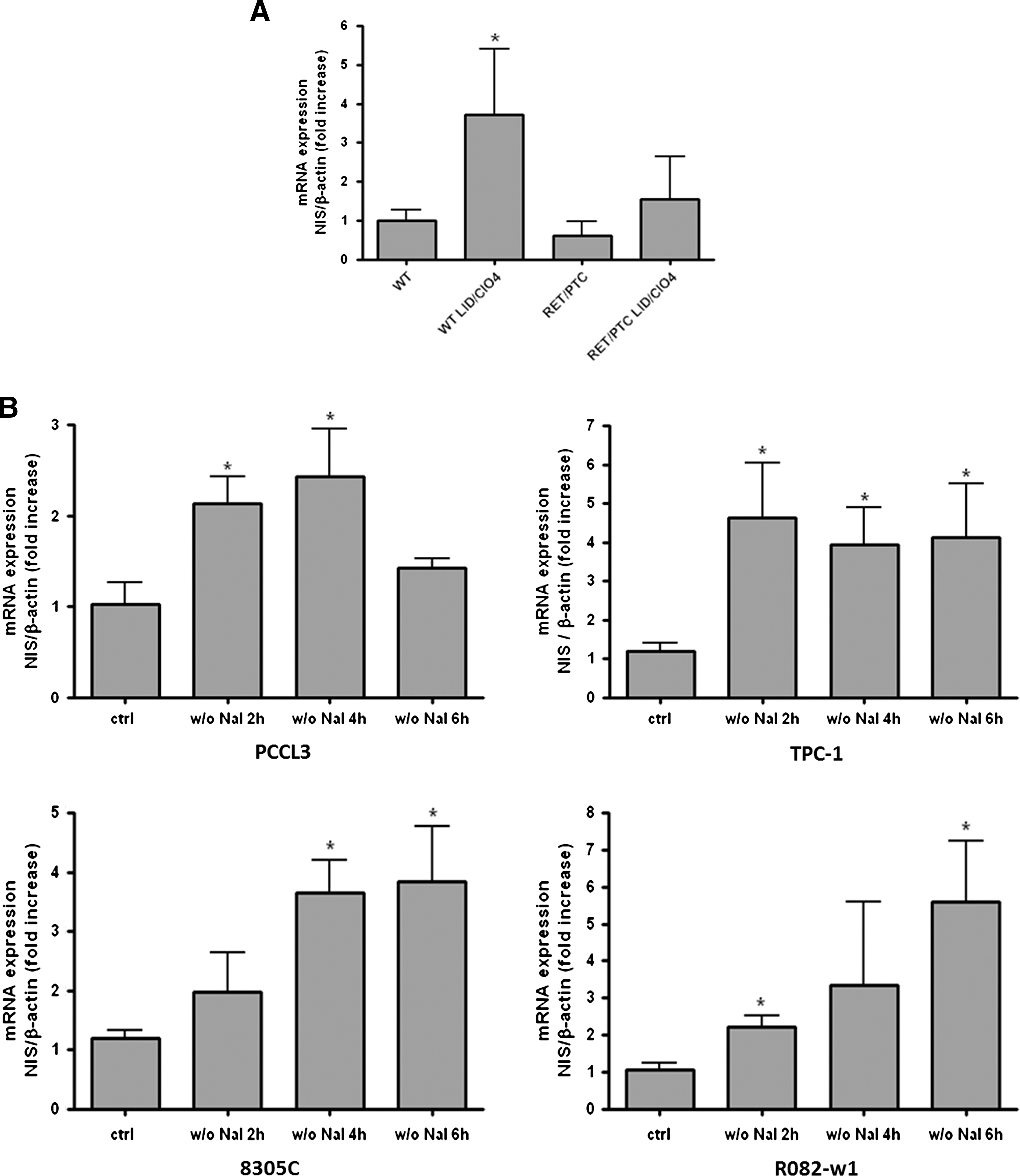
ID-induced NIS mRNA expression in vivo and in vitro.
In the normal PCCL3 cells, TSH concentration being kept steady, ID induced a transient increase in NIS mRNA expression observed after 2 and 4 hours, and then returned to control levels after 6 hours (Fig. 3B).
In the three thyroid carcinoma cell lines that were cultured without TSH, ID also induced an increase in NIS mRNA expression that persisted at least up to 6 hours of ID (Fig. 3B).
ID induces a long lasting VEGF activation in thyroid carcinoma cell lines
As previously shown (19), ID induced a transient increase in VEGF mRNA expression in the normal thyroid cell line PCCL3 reaching a peak at 2 hours that returned back to basal levels after 6 hours (Fig. 4). By contrast, in the three thyroid carcinoma cell lines (TPC-1, 8305C, and R082-w-1), irrespective of the tumor type, VEGF mRNA expression started to increase after 2 hours of ID, and then kept rising up to 6 hours (Fig. 4). These results indicate that, as in normal cells, ID increases VEGF transcription in thyroid cancer cell lines, even though this activation lasts longer, thereby suggesting an absent downregulation.
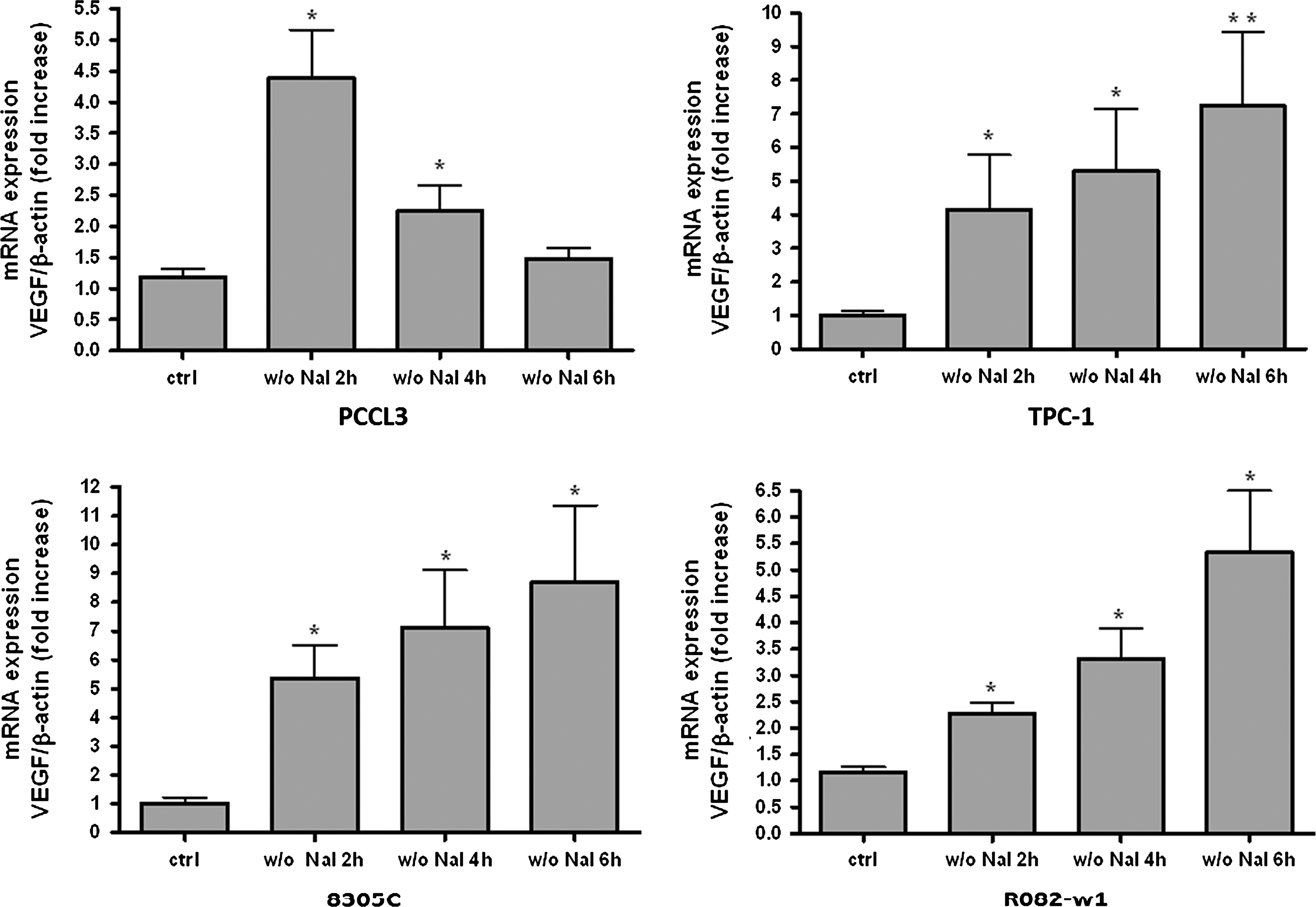
ID-induced increase in the VEGF mRNA expression in normal (PCCL3) and thyroid carcinoma cell lines (TPC-1, 8305C, and R082-w1). Thyroid cells were iodide deprived for 2, 4, or 6 hours. VEGF mRNA levels were measured by qRT-PCR and normalized to β-actin. Values are expressed as mean±SEM of five independent wells (n=5) of one representative experiment. *p<0.05, **p<0.01 as compared with control (ctrl) cells.
ID-induced VEGF is partially dependent on HIF-1 in thyroid carcinoma cell lines
HIF-1 is an important regulator of VEGF (24) that has been previously shown to be implicated in ID-induced VEGF activation in noncancer PCCL3 and in human cells (19). It seemed rational, therefore, to investigate its potential role in regulating VEGF expression in cancer thyroid cells. HIF-1α protein expression was analyzed by western blotting during ID. In the three cell lines, HIF-1α protein expression was slightly but significantly increased after 2 hours of ID, and remained unchanged thereafter for up to 6 hours (Fig. 5).
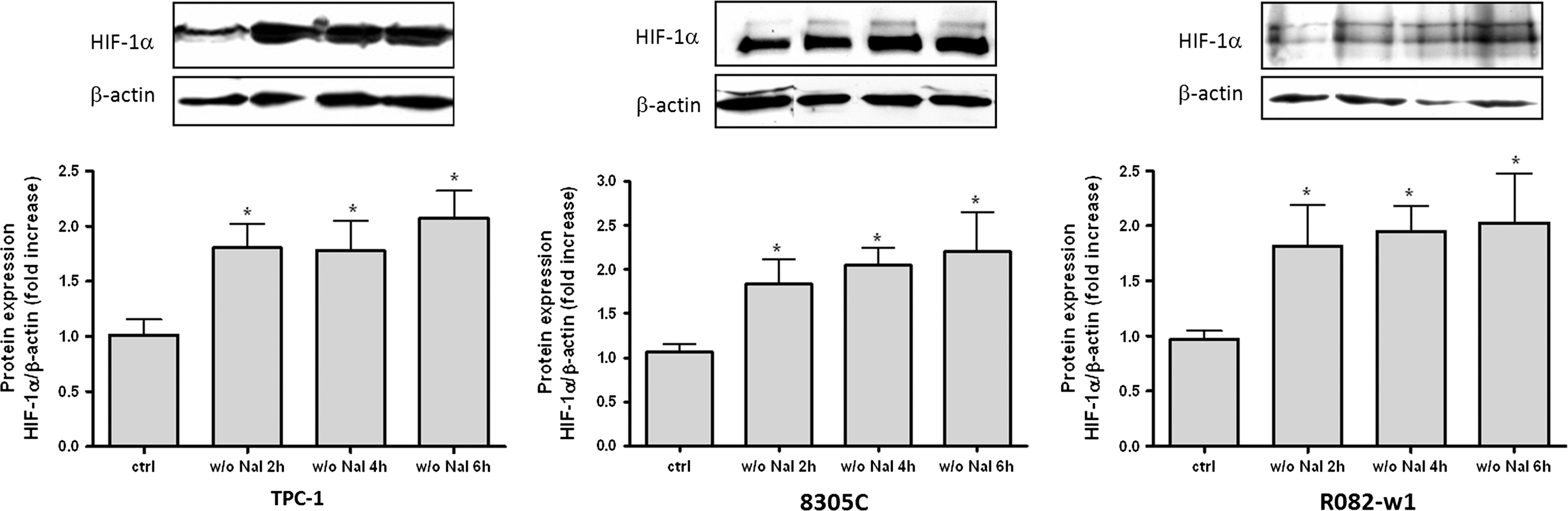
ID-induced increase in hypoxia inducible factor-1α (HIF-1α) protein expression in thyroid cancer cell lines. Thyroid cells were iodide deprived for 2, 4, or 6 hours. HIF-1α protein expression was analyzed by western blot. Densitometric values normalized to β-actin are expressed as mean±SEM of five independent wells (n=5) of one representative experiment. *p<0.05 as compared with control cells.
To test the link between HIF-1α stabilization and VEGF, cells were incubated with echinomycin, a substance that inhibits the binding of HIF-1α to HRE, located in the promoter region of the VEGF-A gene (22). Its specificity of action was recently demonstrated as echinomycin induces similar inhibitory effects as HIF-1α siRNA (25). Echinomycin inhibited ID-induced VEGF mRNA expression, partially in TPC-1 cells, and totally in 8305C cells, but not in R082-w1 cells (Fig. 6). These results indicate that, as in noncancer cells, HIF-1 is implicated in ID-induced VEGF gene activation, but only partially and depending on the cell type.

N-acetylcysteine (NAC) and echinomycin effects on ID (4 hours)–induced VEGF mRNA expression in thyroid cancer cell lines. Thyroid cells were iodide deprived for 4 hours and treated with NAC (1 mmol) or echinomycin (echin, 100 nM). VEGF mRNA levels were measured by qRT-PCR and normalized to β-actin. Values are expressed as mean±SEM of five independent wells (n=5) of one representative experiment. *p<0.05, **p<0.01 as compared with control cells; + p<0.05 as compared with 4-hour iodide-deprived cells.
ID-induced VEGF is ROS independent
Because ID-dependent HIF-1α stabilization is triggered by ROS in normal cells (19), the role of ROS was studied using NAC-induced ROS inhibition in the three thyroid cancer cell lines. NAC treatment had no effect on ID-induced VEGF mRNA expression in both TPC-1 and 8305C cells, while VEGF transcription was actually stimulated in R082-w1 cells (Fig. 6). In the three cell lines, ID-induced HIF-1α protein expression was not significantly altered by NAC (Fig. 7). These results indicate that, in contrast to normal cells, ROS are not part of the intracellular coupling between ID and VEGF activation in the three thyroid cancer cell lines.
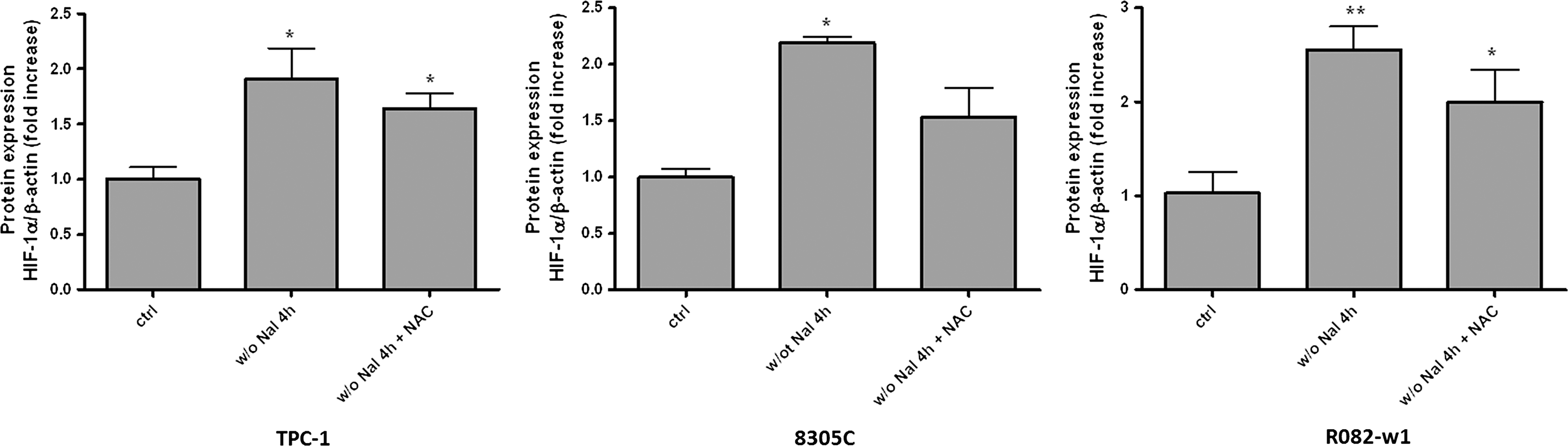
NAC effects on ID (4 hours)–induced increase in HIF-1α protein expression in thyroid cancer cell lines. Thyroid cells were iodide deprived for 4 hours and treated with NAC (1 nM). HIF-1α protein expression was analyzed by western blot. Densitometric values normalized to β-actin are expressed as mean±SEM of five independent wells (n=5) of one representative experiment. *p<0.05, **p<0.01 as compared with control cells.
Discussion
During acute ID, the normal thyrocyte transmits an angiogenic signal, identified as VEGF, to its adjacent endothelial cells. The sudden drop in intracellular iodine stores leads to ROS-mediated stabilization of HIF-1α that in turn activates VEGF. This TSH-independent ROS-HIF-VEGF pathway is triggered in response to ID in the early phase of goiter development to improve the local clearance of iodine, thereby optimizing the functional performance of thyrocytes (18,19). In this work, we report that ID also promotes angiogenesis and VEGF expression in thyroid cancer cells, but via intracellular pathways different than those activated in noncancer cells and not as yet identified. This process might trigger an angiogenic switch in resting in situ avascular microcancers, thereby facilitating tumor progression.
We previously reported that in vitro chronic ID does not influence NIS expression (19). In this work, we show that an acute ID induces a clear upregulation of NIS expression that is observed in vivo (at least in noncancer tissues), as well as in vitro, in both normal and cancer cells. This upregulation was associated with changes neither in circulating TSH in vivo, nor in TSH contents, at least in PCCL3 cells. The absence of TSH influence was even more obvious in cancer cell lines, as they were cultured without TSH. Previous studies reported that in fetuses, NIS expression is also upregulated in case of mild ID without TSH stimulation (26) and that, by contrast, iodine in excess downregulates NIS both in vivo and in vitro (27,28). This downregulation of NIS by iodine in excess is mediated by posttranslational modifications including polyadenylation and mRNA destabilization (29). Conversely, the mechanism underlying the regulation of NIS by ID remains unknown and requires further clarification. It is maybe due to increased sensitivity of thyrocytes to TSH (at least in cells incubated with TSH), but it is neither proved nor explained. Our sole conviction is that ROS are not involved, as our unpublished results indicate that ID-induced NIS mRNA expression is unaffected by NAC, even though basal NIS expression is decreased by ROS, as previously reported (30).
Epidemiological data show that the incidence of thyroid cancers is high in ID areas (2 –4). Nevertheless, while a link between TSH stimulation and cancer development has been shown (8,9), nothing is known about how mild or transient ID, not associated with increases in circulating TSH, impacts thyroid cancer. The hypothesis of this work lies on the idea that ID, independently of increased TSH plasma levels, could per se induce an angiogenic reaction in cancer tissues which, in contrast with noncancer tissues, would run uncontrolled. As a first step, it was therefore important to show that ID actually causes an early angiogenic reaction in cancer thyroid tissues as it does in noncancer tissues. This information was obtained in the in vivo model of RET/PTC3 mice that showed that ID does increase the vascular flow, as well as VEGF gene and protein expression. This result was important as it made it relevant to study of whether and how VEGF is activated by ID in thyroid cancer cells, bearing in mind that tumors, including thyroid cancers, are usually vascularized lesions where VEGF is highly expressed and associated with tumor aggressiveness (31 –34). The tumor growth usually starts from avascular in situ microscopic lesions that may remain quiescent for months to years. Then, for further growth, an angiogenic switch must occur allowing the recruitment of microvessels. The development of tumors also depends on their capacity to adapt to hypoxia and to keep promoting angiogenesis. Angiogenesis is governed by a balance between pro- and antiangiogenic factors and by the capacity of the host microcirculation to respond to these signals (35,36). Many barriers in resting endothelial cells must be overcome, including interactions with adjacent pericytes that keep the endothelium in a nonproliferating state (37 –40). Thus, by inducing an angiogenic phenotype, that is, by increasing the production of VEGF, ID might contribute to the angiogenic switch that triggers the growth of microscopic dormant cancer lesions. The hypothesis that ID may promote recruitment of host microvessels, as well as their activation, is not only supported by our present results, but also by previous results showing that ID induces pericyte detachment and the activation of endothelial cells in goitrous mice (18). Thus, our results support the concept that by triggering an angiogenic switch, in both normal and cancer thyroids, ID may contribute to the progression of tumors, either benign in the case of a physiological adaptation to optimize the functional yield of the gland, or malignant in the case of cancer cells. It is important to note that our present hypothesis does not exclude the recognized role of TSH in tumorigenesis (8,9). It is even likely that both factors are complementary as the second (high TSH plasma levels) often results from the first (ID).
The transcription factor HIF-1 is one of the main regulators of VEGF and plays an important role in tumor development (24). In noncancer thyroid cells, HIF-1 has been shown to mediate ID-induced VEGF activation. The thyrocyte actually translates the acute drop in intracellular iodine into a signal that then stabilizes HIF-1α protein via ROS (19). During cancer growth, some cells may become hypoxic, which will activate various HIF-1-responsive genes, including VEGF, GLUT-1, and carbonic anhydrase IX (CA-9). The analysis of human thyroid carcinomas has confirmed that the level of HIF-1α protein expression is usually higher than in benign thyroids (41 –43). In the thyroid cancer cells that we analyzed, ID also induces HIF-1α protein, but likely with different cellular consequences than those observed in noncancer cells. It is well illustrated by our observation that echinomycin differentially impacts VEGF transcription in different types of cancer cells. For instance, in papillary carcinoma cells (TPC-1 cells), VEGF gene activation was only partially inhibited by echinomycin, but fully inhibited in undifferentiated carcinoma (8305C cells), and not at all in follicular carcinoma (R082-w1 cells). As HIF-1α expression in thyroid cancers is very heterogeneous, being more pronounced in invasive zones and in anaplastic carcinomas (41,42), these results suggest that the influence of HIF-1α may be variable depending on the type of cancer. Thus, in more aggressive cancers, like undifferentiated carcinomas, the angiogenic phenotype would depend more on HIF-1 than in differentiated types. In addition, pathways other than HIF-1 are likely involved in ID-induced VEGF gene activation. Among candidates, one may consider the activation of VEGF by IGF-1 via a PI3K/Akt-dependent pathway (44), or the involvement of an endoplasmic reticulum stress where VEGF is activated independently from HIF-1, but in relation with unfolded protein response-associated transcription factors, such as ATF4 and XBP1(S) (45,46). Our data are therefore in line with the paradigm that alternative intracellular pathways likely not as tightly controlled as in noncancer cells are activated in cancer cells, making them ready to release angiogenic factors, such as VEGF, potent enough to induce a pathologic angiogenic switch.
In conclusion, we propose that a link exists between ID and thyroid cancers. By favoring an angiogenic phenotype in thyroid cancer cells, ID might provide a favorable environment for tumor growth. ID induces VEGF transcription in thyroid cancer cells via intracellular pathways partially depending from HIF-1 as in noncancer cells, but also via other pathways likely not as well controlled as in noncancer cells, which may infer for the likelihood of anarchic proliferation.
Footnotes
Acknowledgments
This work was supported by the Fonds National pour la Recherche Scientifique (F.R.S.-FNRS), and the Fonds de la Recherche Scientifique Médicale (FNRS-FRSM) and by the FP6 (GENRISK-T, FIP6-2006-036495) and FP7 (DoReMi, agreement n.o. 249689) EURATOM projects. P.S. is an F.R.S.-FNRS research associate.
Disclosure Statement
The authors declare that no conflict of interest exists.