
Abstract
Background:
Riedel's thyroiditis (RT) is a very rare chronic fibrosing disorder of unknown etiology that is often associated with multifocal fibrosclerosis (MFS). Immunoglobulin (Ig) G4–related sclerosing disease (IgG4-RSD), a new clinico-pathological entity also associated with MFS, is characterized by IgG4+ plasma cell infiltration and fibrosis in one or more organs. Although the association of RT and IgG4-RSD has been suggested, it has seldom been studied or reported. We report a classical case of RT with serological (IgG4 levels) and immunohistochemical (IgG and IgG4) assessment, in search of an underlying IgG4-RSD.
Patient:
The patient was a 57-year-old woman who underwent a subtotal thyroidectomy for a long-standing goiter with a rapidly enlarging isthmic nodule.
Results:
Histopathological examination of the surgical specimen revealed all of the morphological features of RT and IgG4-RSD, including partial fibrosis of the thyroid gland with destruction of the thyroid follicular architecture; obliterative phlebitis; and a mixed infiltrate composed of lymphocytes, eosinophils, and plasma cells. The fibro-inflammatory process extended beyond the thyroid capsule into the surrounding tissues. Immunohistochemical examination revealed approximately 70 IgG4+ plasma cells per high power field (HPF) with an IgG4/IgG ratio of 35%. Although serum levels of IgG4 were normal (20 mg/dL), total IgG levels were slightly elevated (1370 mg/dL). There was no evidence of involvement of other organs at the time of RT diagnosis.
Conclusions:
The morphological similarities between RT and IgG4-RSD suggest that these entities are closely related. Therefore, RT with increased IgG4+ plasma cells, with or without elevated IgG4 serum levels, may represent the first clinical manifestation of an underlying IgG4-RSD. However, due to the rarity of both conditions and the limited specificity and sensitivity of both IgG4 serum levels and IgG/IgG4 immunohistochemistry in the diagnosis of IgG4-RSD, further studies are needed to verify this hypothesis.
Introduction
Although the cause of RT remains unknown, an association between RT and autoimmunity has been observed (2). Since they often occur in the same patient, many authors have suggested that RT and similar disorders, such as retroperitoneal or mediastinal fibrosis, sclerosing pancreatitis, and orbital pseudotumors, are manifestations of the same underlying systemic disease. Hence, multifocal idiopathic fibrosclerosis (MFS) is among the several terms used to describe this systemic process (1,2). One-third of patients with RT develop fibrosing disorders in other organs over a 10-year period. However, of all the MFS cases, RT is present in a much smaller subset of patients (1). RT can also be associated with HT or Graves' disease (2). While RT has no standardized treatment, most patients are treated with corticosteroids and/or tamoxifen (2).
Over the last few years, a new clinico-pathological entity titled Immunoglobulin (Ig) G4–related sclerosing disease (IgG4-RSD) has been characterized (5). This syndrome predominantly affects middle-aged and elderly men. Patients generally present with symptoms referable to the involvement of one or more sites, usually in the form of mass lesions. They usually have a good general condition, with no fever or constitutional symptoms. The prototype for this condition is sclerosing or autoimmune pancreatitis, which most commonly presents as painless obstructive jaundice with or without a pancreatic mass. Other common sites of involvement are the hepatobiliary tract, salivary gland, orbit, and lymph node; however, practically any organ can be affected, and the list of organs affected continues to grow. Common laboratory findings include increased serum globulin, IgG, IgG4, and IgE levels. The natural history of the disease is characterized by the development of multiple sites of involvement over time, sometimes after many years. However, IgG4-RSD occasionally remains localized to one site. The disease often responds well to steroid therapy. The main pathologic findings in various extranodal sites are morphologically indistinguishable from RT and include lymphoplasmacytic infiltration, sclerosis, obliterative phlebitis, and the atrophy and loss of the specialized structures of the involved tissue. Immunostaining shows increased numbers of IgG4+ plasma cells in the involved tissues. To date, the cut-off number of IgG4+ plasma cells for positive diagnosis has not been standardized. To diagnose IgG4-RSD, Cheuk and Chan (5) recommends both an absolute number of IgG4+ plasma cells (>50 per high power field (HPF), corresponding to the average number of IgG4+plasma cells in three HPF from the areas with the highest IgG4+ plasma cell density) and a ratio of IgG4+/IgG+ cells >40%. The total number and proportion of IgG4+ plasma cells is more sensitive than serum IgG4 titers, which are not elevated in ∼30% of cases but in contrast are mildly elevated in 7%–10% of patients with pancreatic carcinoma. However, both elevated numbers of IgG4+ plasma cells and IgG4/IgG ratios have been described in various localized non-specific chronic inflammatory conditions, including the reactive fibro-inflammatory response associated with pancreatic carcinoma (6).
Since MFS has been shown to represent IgG4-RSD in a significant proportion of cases (4), it has been assumed that RT, a known component of MFS, also belongs to this disease spectrum. However, due to the rarity of RT and the recent characterization of IgG4-RSD, very few data are available in the literature to verify this hypothesis. Here, we describe a classical case of RT in a 57-year-old female patient who was assessed serologically (IgG4 levels) and immunohistochemically (IgG and IgG4) for an underlying IgG4-RSD.
Methods
The subtotal thyroidectomy surgical specimen was fixed in 10% buffered formalin and routinely processed. The specimen was extensively sampled. Hematoxylin and eosin, trichrome, and immunohistochemical stains were performed on 3-μm-thick sections of formalin-fixed, paraffin-embedded tissue.
Immunohistochemistry was conducted using the following antibodies and conditions: anti-CD 20 (Dako, Carpinteria, CA; clone L26, 1:100 dilution), anti-CD3 (Dako; clone F7.2.38, 1:20 dilution), anti-CD5 (Novocastra, Newcastle on Tyne, United Kingdom; clone 4C7, 1:50 dilution), anti-CD138 (Dako; clone MI-15, 1:20 dilution), anti-MUM-1 (Dako; clone MUM-1p, 1:20 dilution); anti-kappa light chain (Dako; polyclonal, 1:30,000 dilution), anti-lambda light chain (Dako; polyclonal), anti-IgG (Dako; polyclonal, 1:20,000 dilution), anti-IgA (Dako; polyclonal, 1:6000 dilution), and anti-IgG4 (Invitrogen, Carlsbad, CA; clone HP6025, 1:50 dilution). Antigen retrieval was performed using antigen retrieval buffer (100× Tris-ethylenediaminetetraacetic acid buffer, pH 9.0) for staining for CD20, CD3, CD5, CD138, and MUM-1; microwave pretreatment for 10 minutes was performed before staining for IgG4 and kappa and lambda light chains. Trypsin and proteinase K pretreatments were performed before staining for IgG and IgA, respectively. The Envision Plus detection system (Dako) was used for kappa, lambda, IgG, IgA, and IgG4 staining.
The areas with the highest density of IgG4+ plasma cells from three slides were selected. The total number of IgG4+ plasma cells was counted in three HPF of the selected areas; counting was performed at a magnification of 400×, and the HPF area diameter was 0.52 mm. The mean number of IgG4+ plasma cells was then calculated.
Results
Clinical history
A 57-year-old woman known to have a long-standing goiter presented subclinical hyperthyroidism and a rapidly growing isthmic nodule that grew up to 4 cm over ∼6 months. Thyroid function tests were as follows: thyroid-stimulating hormone 0.13 mIU/L (reference range [RR] 0.3–4.3), free thyroxine 13.2 pmol/L (RR 9–19), and free triiodothyronine 5 pmol/L (RR 2.6–5.7). Levels of both thyroglobulin and anti-thyroglobulin antibodies were elevated, 969 μg/L (RR 5–25) and 6.7 kIU/L (RR <4.1) respectively. Palpation revealed a diffusely enlarged thyroid gland with a firm 4 cm isthmic nodule. However, the patient did not present with any local or compressive symptoms. Her previous medical history was unremarkable; she was a non-smoker.
Thyroid ultrasonography showed a bilateral multinodular goiter with a 3.7 cm isthmic nodule. A total thyroidectomy, without previous fine-needle aspiration, was planned. However, during surgery, the thyroid region was found to be largely infiltrated with white, sclerotic intra- and extrathyroidal mass lesions that prevented complete resection. Moreover, the thyroid gland was diffusely firm, on both sides, including both superior poles. An intraoperative pathological examination was performed to exclude a malignancy. The frozen section revealed a fibro-inflammatory process in the perithyroidal tissue without evidence of carcinoma. A bilateral subtotal thyroidectomy was then performed with incomplete resection of the sclerotic mass lesion; a complete resection was not performed to preserve the recurrent laryngeal nerves and the parathyroid glands.
Although total serum IgG levels measured 3 weeks after surgery were slightly elevated (1370 mg/dL; RR, 670–1318 mg/dL), serum IgG4 levels were normal (20 mg/dL; RR, 8.0–140.0 mg/dL). The patient did not receive corticosteroid treatment.
The post-operative course was uneventful except for some bruising around the scar. The stimulation of both vagus nerves at the end of the procedure (neuromonitoring) showed a good response of the vocal folds. The patient did not experience dysphonia, and the calcium and parathormone levels were in the normal range postoperatively. A thoraco-abdominal computed tomography scan, performed 4 months later, did not show any abnormality that was suggestive of MFS.
Pathological examination
The right thyroid lobe and isthmus region of the surgical specimen was fragmented and contained very firm white areas that were predominately located at the periphery of the thyroid parenchyma and involved whole fragments up to the surgical margins. Multiple colloid-like nodules were also present in the thyroid parenchyma, consistent with a multinodular goiter (Fig. 1).

Macroscopic aspect of the surgical specimen (formalin fixed and sliced) showing a dense, white, well-defined area inside and around the multinodular thyroid parenchyma. A square in the background represents an area of 1 cm2.
Microscopically, the thyroid gland was partially replaced by a fibro-inflammatory infiltrate that extended into the perithyroidal soft tissues and musculature (Fig. 2a–c). The inflammatory infiltrate comprised of lymphocytes, plasma cells, and some eosinophils; however, lymphoid follicles were absent. Dense keloid-like sclerotic areas with a few inflammatory cells were also present (Fig. 2b). In addition, numerous veinulitis foci were observed (Fig. 2d).
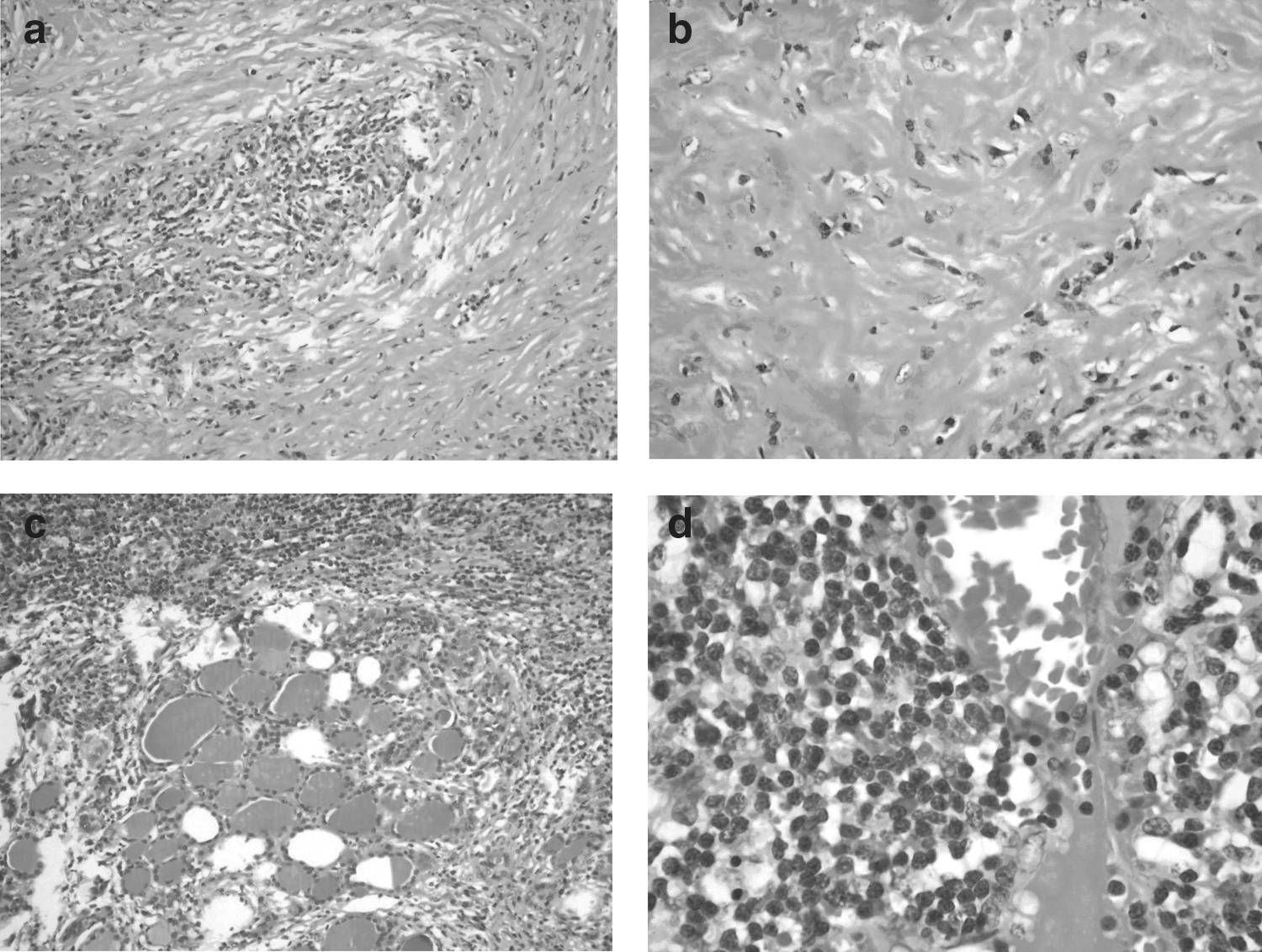
Histopathology of Riedel's thyroiditis case showing
The lymphoid population comprised of T cells (CD3+), B cells (CD20+), and plasmocytes (CD138 and MUM-1+). Kappa and gamma light chain expression was approximately equal. Although plasma cells were predominately IgG+, a small population of IgA+ plasma cells was also observed. There were approximately 70 IgG4+ plasma cells per HPF, with an IgG4/IgG ratio of 35%.
Discussion
This case of RT with increased numbers of IgG4+ plasma cells (70/HPF) and an IgG4/IgG plasma cell ratio of 35% suggests that RT and IgG4-RSD overlap clinically, morphologically, and immunohistochemically and that RT may represent an inaugural manifestation of IgG4-RSD in the thyroid region. Nevertheless, increased serum levels of IgG4 (>135 mg/dl), though not essential for the diagnosis of IgG4-RSD (5), would have further supported the association between these two entities. Although the association between RT and IgG4-RSD has been previously suggested by several authors (2,4,7 –10), very few cases have been closely studied that determine a relationship between these two entities which are morphologically and strikingly similar.
To date, no cases of RT with elevated IgG4 serum levels have been reported (1). This biological feature is critical, because the occurrence of variably high numbers of IgG4+ plasma cells is observed in a number of diverse, non-specific inflammatory conditions (6). Thus, high IgG4+ plasma cell counts and high IgG4/IgG ratios do not always reliably distinguish between IgG4-RSD and non-specific inflammatory conditions. Therefore, IgG4 plasma cell counts should be cautiously interpreted in the context of appropriate clinical and histopathological features (6). For a comparison, we examined a recent case of thyroid papillary carcinoma with severe sclerosis and inflammatory reactions reminiscent of IgG4-RSD. In this specimen, the total number of IgG4+ plasma cells (>100) and the IgG/IgG4 ratio (90%) were both much higher compared with the RT case reported here.
In the largest retrospective study conducted on the available RT (3), only 2 patients out of 21 were assessed (serum IgG4 levels and tissue immunostaining) for an underlying IgG4-RSD. One had low serum IgG4 levels (7.6 mg/dL; reference range, 8.0–140 mg/dL) with normal values for the remaining IgG subclasses. Her thyroid specimen had “a few IgG4+ plasma cells” visible on immunostaining. The other one had normal serum IgG4 levels, and the thyroid tissue was “moderately positive” for IgG4. Based on this limited description and the limited specificity/positive predictive value of immunostaining (as just mentioned), no conclusive association between RT and IgG4-RSD can be inferred from these data.
Jakobiec et al. reported a case of MFS, including lacrimal gland and thyroid (RT) involvement, which was assessed serologically and immunohistochemically (lacrimal gland and thyroid) for an underlying IgG4-RSD (7). Serum IgG4 levels were normal, and the tissue density of IgG4+ plasma cells was low. However, these results may have been influenced by the administration of prednisone, as IgG4-RSD is known to be corticosteroid responsive. Finally, Dahlgren et al. retrospectively studied three cases of RT with immunohistochemistry in search of an association with IgG4-RSD (4). The authors concluded that RT is a part of the IgG4-RSD spectrum; however, they used a lower threshold for diagnosis (either >50% IgG4/IgG ratio or >10 IgG4+ plasma cells/HPF) than those recommended by Cheuk and Chan (>50% IgG4/IgG ratio and >50 IgG4+ plasma cells/HPF). In addition, serum IgG4 levels were available only in one patient and were normal. Although IgG4+ plasma cells were present in all cases, the total and relative (IgG4/IgG) amounts were variable. In the thyroid parenchyma, 2 out of 3 patients had elevated IgG4/IgG ratios (>40%). Of these two patients, one had >10 IgG4+ plasma cells/HPF. Interestingly, the other patient also exhibited extensive extrathyroidal disease, including cervical lymph node involvement (with 110 IgG4+ plasma cells and an IgG4/IgG ratio of 0.97%), that was diagnostic of MFS and IgG4-RSD. The third patient had neither significantly elevated serum IgG4 levels nor an increased IgG4/IgG ratio.
Recent studies have also suggested a relationship between HT and IgG4-RSD and described a unique subtype of HT called immunoglobulin IgG4-thyroiditis, which is characterized by lymphoplasmacytic infiltration, fibrosis, increased numbers of IgG4+ plasma cells in the thyroid, and high serum IgG4 levels. From a clinical aspect, IgG4-thyroiditis presents significantly different characteristics compared with non-IgG4-thyroiditis (8 –10). Significantly, the immunohistochemical threshold used in these studies was also lower (>20 IgG4+ plasma cells/HPF and >30% IgG4/IgG ratio) compared with the diagnostic criteria recommended by Cheuk and Chan (5). Although RT and the fibrosing variant of HT were once considered morphologic variants of the same disease, these diseases have been considered distinct clinico-pathologic entities since the 1970s (11). Currently, some authors have suggested that RT and the fibrosing variant of HT are, respectively, a diffuse or localized manifestation of IgG4-RSD (9,10). However, further investigation is needed to clarify the potential link between these entities. HT, including its fibrosing variant, and RT may represent different clinical, spatial, and/or temporal manifestations of the same underlying condition, that is, IgG4-RSD, whose natural course may be influenced by the treatment (e.g., surgery, corticosteroids). Indeed, the diagnosis of IgG4-RSD early in its onset is important, as adequate follow-up and treatment may eventually prevent other organs from being affected.
In conclusion, the clinical, morphological, and immunohistochemical overlap between RT and IgG4-RSD, as illustrated by this case, suggests that these entities are closely related. Therefore, RT cases with elevated numbers of IgG4+ plasma cells, with or without elevated IgG4 serum levels, may represent the first clinical manifestation of an underlying case of IgG4-RSD; furthermore, RT should be added to the list of entities that are potentially associated with IgG4-RSD. Diagnostic work-up of RT cases should ideally include immunohistochemical plasma cell assessment (IgG and IgG4) and circulating IgG4 levels. However, more data are needed to clarify the link between these two entities and to identify more specific diagnostic criteria.
Footnotes
Disclosure Statement
The authors disclose that no competing financial interests exist.