
Abstract
Background:
Thyroid hormone (TH) plays an important role in the modulation of cardiac function, including contractility and systemic vascular resistance (SVR). 3,5,3′-triiodothyronine (T3), the active form of TH, induces the activation of endothelial nitric oxide synthase via PI3K/AKT non-genomic signaling. Hypothyroidism is associated with an increase in SVR and serum low-density lipoproteins (LDL) levels, and accumulation of oxidized LDL (oxLDL) may impair endothelial-dependent vascular relaxation. The aim of this study was to investigate the effects of both native LDL (nLDL) and oxLDL on T3-mediated AKT phosphorylation, nitric oxide (NO), and cyclic guanosine monophosphate (cGMP) production in human endothelial cells.
Methods:
Human umbilical vein endothelial cells were exposed to either nLDL or oxLDL for 3 hours and then stimulated with T3 (10−7 M) or pretreated with an antioxidant mixture of vitamins E and C for 12 hours before treatment with LDL. An analysis of AKT phosphorylation was performed by Western blot, and NO production was evaluated by using 4,5-diaminofluorescein diacetate. Intracellular production of cGMP was measured by enzymatic immunoassay. LDL oxidation was carried out by incubating LDL with CuSO4, and α-tocopherol content of LDL was evaluated by high-performance liquid chromatography.
Results:
OxLDL impaired T3-mediated AKT phosphorylation at serine 473 and significantly decreased the production of both NO (oxLDL+T3 vs. T3, 9.79±0.5 AU vs. 80.75±2.8 AU, mean±standard deviation, p<0.0001) and cGMP. Furthermore, pretreatment with the antioxidant mixture obviated the inhibitory effect of LDL on T3 action.
Conclusions:
The results of this study demonstrate that oxLDL may contribute to a blunting of the non-genomic action of T3 and impair the effect of T3 on NO and cGMP production in endothelial cells. These data suggest that oxLDL, apart from inducing the atherosclerotic process, may also promote a mechanism of peripheral resistance to T3, further amplifying the impact of hypothyroidism on endothelial function by increasing SVR.
Introduction
Materials and Methods
Cell culture and reagents
Human umbilical vein endothelial cells (HUVECs; Lonza) were maintained in a complete endothelial growth medium containing 2% fetal bovine serum (FBS), recombinant human vascular endothelial growth factor (VEGF), basic fibroblastic growth factor, human epidermal growth factor, insulin-like growth factor 1, hydrocortisone, ascorbic acid, heparin, and GA-1000 (gentamicin and amphotericin B, 1 μg/mL), according to the instructions of the supplier (EGM-2 bullet kit; Lonza). HUVECs were used between passages 3 and 6 and grown in 5% CO2 at 37°C. Before each experiment, cells were plated on 100 mm dishes or in Labteck chamber slides (Nunc) and starved overnight in phenol red-free endothelial basal medium (EBM) with 1% dialyzed FBS (Gibco) before treatment. Vitamin E, vitamin C, Wortmannin, T3, and lyophilized LDL from human plasma were purchased from Sigma-Aldrich. A stock solution of 20 μg/mL of T3 was prepared by dissolving 1 mg of T3 in 1 mL of 1-normality (N) NaOH, and brought to a final concentration of 20 μg/mL (30 μM) by adding 49 mL of sterile medium. Aliquots were stored at −20°C. This T3 stock solution was further diluted in medium immediately before use, and the amount of NaOH was normalized in each sample.
LDL oxidation and vitamin E estimation
LDL oxidation was carried out by incubating LDL (50 μg protein/mL) at 37°C for one hour with 5 μM CuSO4 solution. Both nLDL and oxLDL were dialyzed overnight, and the degree of LDL oxidation was monitored as previously described (17). For α-tocopherol estimation, tocopheryl acetate (internal standard) was added to 100 μL of LDL. The mixture was deproteinized with an equal volume of ethanol and then extracted twice with hexane. Phase separation was achieved by centrifugation. The collected upper phase was evaporated under nitrogen, and samples were analyzed by high-performance liquid chromatography (HPLC) (18). α-Tocopherol content was expressed as μg/50 μg protein/mL of LDL. Samples were dissolved in 100 μL of methanol:diethyl ether (4:1), and 50 μL were injected in an ODS-C18 Spherisorb column (250 mm×4 mm, 5 μm particle size). The mobile phase was a mixture of methanol:water (99:1, v/v) at a flow rate of 1 mL/min. Absorbance was monitored at 290 nm using an LC-235 Diode Array Detector (Perkin-Elmer), as described by Ortega et al. (19,20).
Western blot analysis
HUVECs were grown in 100-mm dishes, and starved overnight in phenol-red free EBM containing 1% dialyzed serum. Cells were then treated with T3 (10−7 M) for 10, 20, 30, and 60 minutes for time course experiments, or treated with oxLDL (50 μg protein/mL) or nLDL (50 μg protein/mL) for 3 hours followed by T3 (10−7 M) stimulation for 20 minutes. When indicated, cells were pretreated for 12 hours with a mixture of vitamin E (150 μM) and vitamin C (150 μM); then incubated with either LDL (50 μg/mL) or oxLDL (50 μg/mL) for 3 hours; and stimulated with T3 (10−7 M) for 20 minutes. Cells were then washed with cold phosphate-buffered saline, harvested, and cell lysates were prepared in RIPA buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid, 1% Triton, and 0.1% sodium dodecyl sulfate), containing protease inhibitors cocktail (1% v/v), phosphatase inhibitors cocktail (1% v/v) (Sigma-Aldrich), and 20 mM N-ethylmaleimide (Sigma-Aldrich), for 40 minutes at 4°C. Cell lysates were centrifuged for 10 minutes at 12,000 g at 4°C, and the supernatants were collected. Cell lysates were analyzed for protein content by using Bradford assay with bovine serum albumin as a standard. Lysates were then mixed with 4×lithium dodecyl sulfate (LDS) buffer (Invitrogen) and heated for 10 minutes at 70°C. Total cell lysates (60 μg of proteins) were separated by 4%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (NuPAGE; Invitrogen), transferred to a nitrocellulose membrane, and blocked in 5% non-fat dry milk in Tween-TBS. The nitrocellulose was incubated overnight at 4°C with different primary antibodies and then treated with specific horseradish peroxidase-conjugated antirabbit or antimouse secondary antibodies. The filters were developed by enhanced chemiluminescence using Kodak X-Omat film. The primary antibodies used were as follows: rabbit anti phospho-AKT (serine 473; Abcam), rabbit anti phospho-eNOS (serine 1177; Abcam), and mouse monoclonal anti–β-actin (Cell Signaling Technology). Densitometric measurements of the bands in Western blot analysis were evaluated using Quantity One software (Bio-Rad).
Measurement of NO production
Intracellular NO production was assessed using the NO-specific fluorescent dye 4,5-diaminofluorescein diacetate (DAF-2 DA; Cayman Chemical). HUVECs were grown in complete medium in Labteck chamber slides (Nunc) and starved overnight in phenol red-free EBM containing 1% dialyzed FBS. Cells were treated for 20 minutes with T3 (10−7) or pretreated for 3 hours with oxLDL (50 μg/mL) or nLDL (50 μg/mL) and then stimulated with T3 (10−7). When indicated, cells were pretreated for 12 hours with the antioxidant mixture of vitamins C and E. DAF-2 DA was added to the cell culture medium (final concentration 3 μM) for 20 minutes at 37°C. Cells were rinsed twice with phenol red-free EBM and fixed in 2% paraformaldehyde for 20 minutes at 4°C. Fixed cells were examined with an Olympus BX51 microscope, using appropriate filters with a peak excitation wavelength of 480 nm and a peak emission wavelength of 510 nm. Images were captured using IP Labs Software (Scanalytics, Inc.) and analyzed using ImageJ 1.40g (Wayne Rasband, National Institute of Health).
cGMP assay
cGMP production was assessed with the aim of evaluating the effect of oxLDL and nLDL on T3-mediated, NO-dependent cGMP production. HUVECs were grown in 100 mm dishes in complete medium to 85% confluence and starved overnight in phenol red-free EBM containing 1% dialyzed FBS. Cells were treated for 20 minutes with T3 (10−7 M) or pretreated for 3 hours with oxLDL (50 μg/mL) or nLDL (50 μg/mL) and then stimulated with T3 (10−7 M). When indicated, cells were previously exposed for 12 hours to the antioxidants. The medium was then removed, and the cells were immediately covered with 0.1 M HCl and incubated at room temperature for 20 minutes before harvesting. The HCl extract was collected, centrifuged for 10 minutes at 1000 g, and the direct measurement of the intracellular cGMP levels was performed using the cGMP direct EIA Kit (Cayman) according to the manufacturer's instructions. Protein content in each plate was measured by the Bradford assay to normalize cGMP values.
Statistical analysis
All the data are reported as mean±standard deviation and are representative of three independent experiments. Statistical analysis was performed using a two-tailed Student's t-test using Sigma Plot software (Systat Software, Inc.). The difference between the control and treated groups was considered statistically significant at p<0.05.
Results
oxLDL inhibits T3-mediated AKT phosphorylation in endothelial cells
To determine whether oxLDL can affect T3-mediated NO production in vascular endothelium, we first assessed the effect of T3 on AKT and eNOS phosphorylation. To this end, HUVECs were treated with or without T3 (10−7 M) at different time points. Western blot analysis showed a significant time-dependent increase of AKT phosphorylation at serine 473 and eNOS phosphorylation at serine 1177 with maximal activation levels at 20 and 60 minutes, respectively (Fig. 1). Since eNOS is a downstream target of AKT, these data are in keeping with the stimulatory effect of T3 on this pathway.
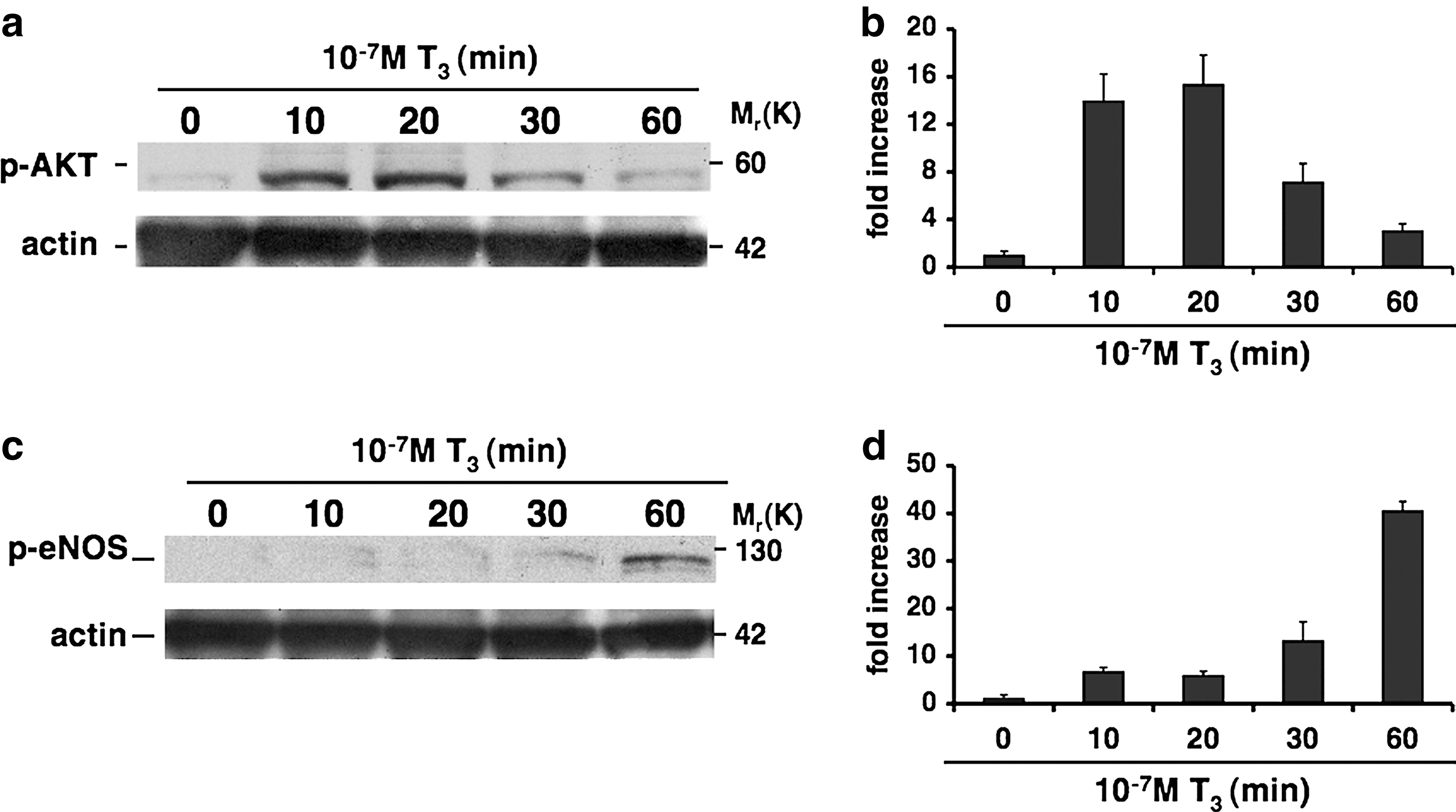
Effects of 3,5,3′-triiodothyronine (T3) on AKT and endothelial nitric oxide synthase (eNOS) phosphorylation. Human umbilical vein endothelial cells (HUVECs) were treated with or without T3 (10−7 M) at different times as described in Materials and Methods. Equal amounts of protein were then resolved on 4%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto a nitrocellulose membrane, probed with appropriate primary and secondary antibodies, and visualized by enhanced chemiluminescence. A representative western blot from three independent experiments indicates the increase of AKT phosphorylation at serine 473 and eNOS phosphorylation at serine1177 with different fold increases that were time dependent, with maximal activation levels at 20 minutes for phospho-AKT serine 473
Since LDL play a crucial role in the early stage of atherosclerosis, as well as in NO availability and metabolism, we next tested whether LDL could play a role in modulating the non-genomic activity of T3. Hence, we investigated the effects of both nLDL (50 μg/mL) and oxLDL (50 μg/mL) on T3-mediated AKT phosphorylation.
LDL oxidation was carried out by incubating LDL with CuSO4 solution, and α-tocopherol content in LDL was performed as described in Materials and Methods (Fig. 2a, b). Cells were exposed to either oxLDL or nLDL for 3 hours followed by an exposure to T3. In order to test the PI3K/AKT pathway inhibition, cells were either untreated or pretreated with Wortmannin (10−7 M) for 30 minutes, and then stimulated with T3 for 20 minutes. Western blot analysis showed the inhibition of AKT phosphorylation in the cells treated with both forms of LDL, mimicking the inhibitory effect of Wortmannin (Fig. 2c, e).
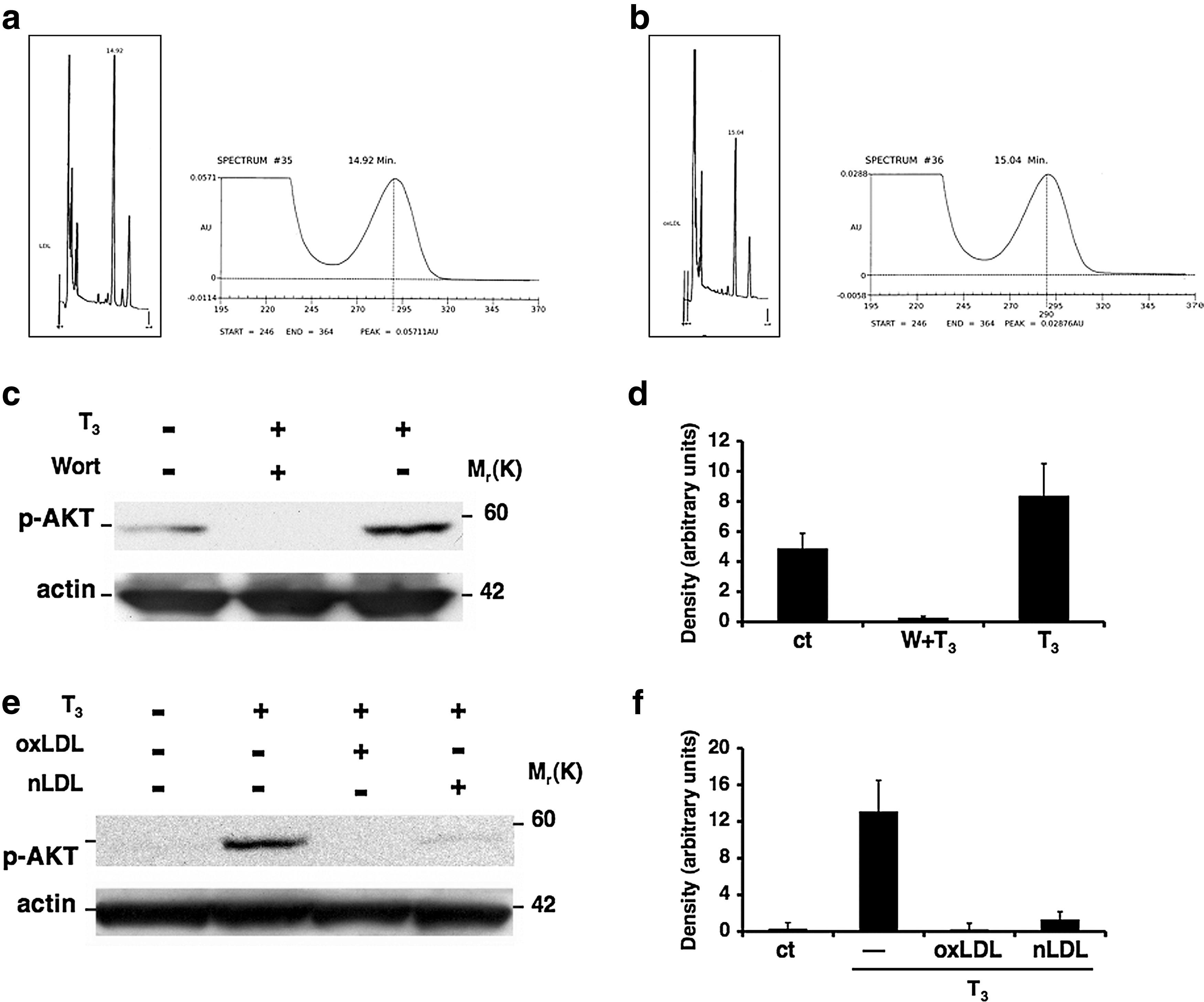
Inhibition of T3-mediated AKT phosphorylation by oxidized low-density lipoproteins (oxLDL). High-performance liquid chromatography of α-tocopherol content of LDL. Chromatogram of α-tocopherol content of native LDL (nLDL; non oxidized) corresponding to 48.46 μM
We, thus, hypothesized that during the cell treatments with LDL, the native form of LDL underwent oxidation, leading to the same result obtained as with oxLDL treatment. To support this hypothesis, the cells were pretreated with an antioxidant combination of vitamin E and vitamin C. The data clearly show that this treatment prevented the inhibitory effect of both nLDL and oxLDL on AKT phosphorylation (Fig. 3a), indicating that the oxidation of LDL was causing the inhibition of T3-mediated AKT phosphorylation.
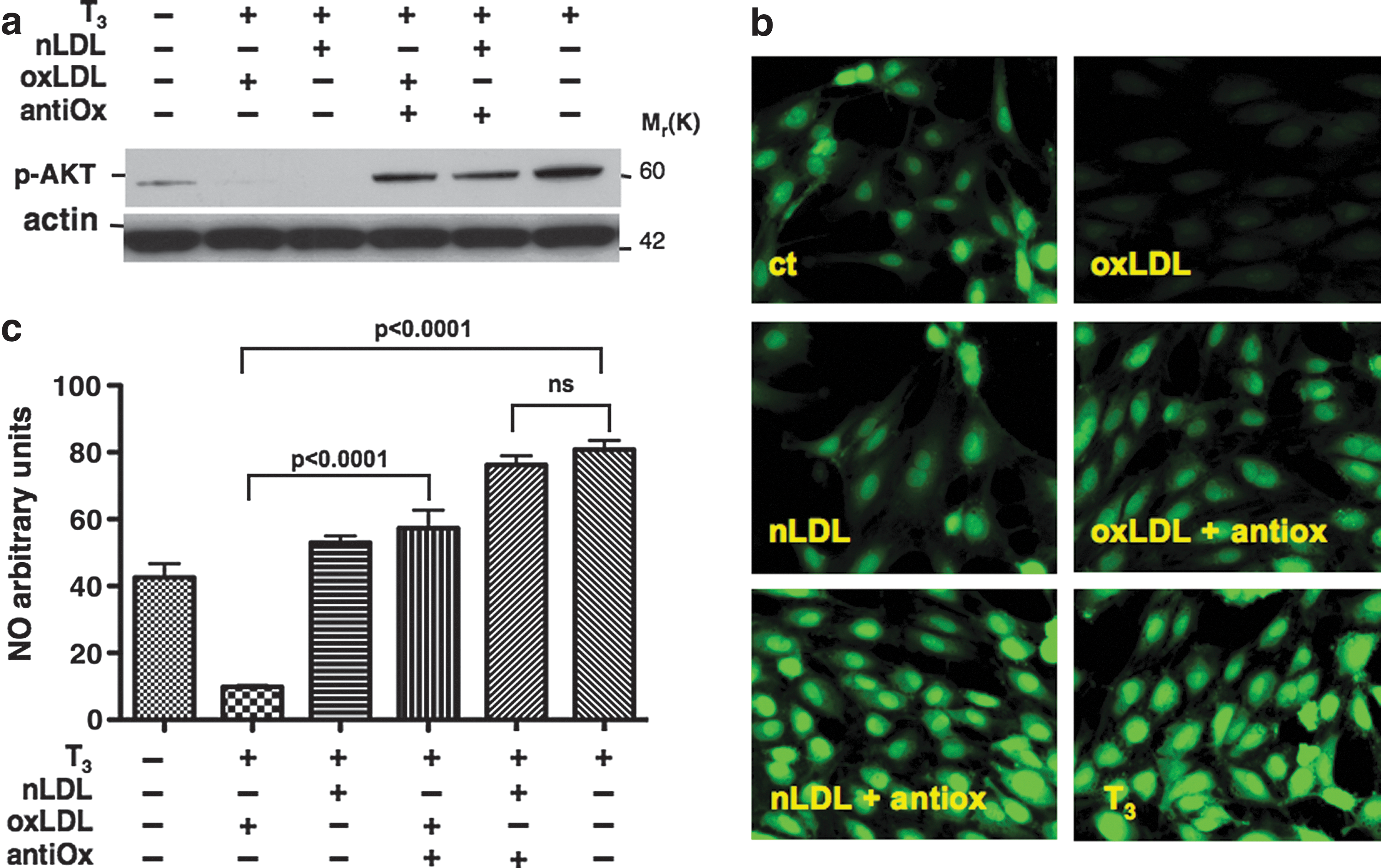
oxLDL impair T3-induced nitric oxide (NO) production. Antioxidant mixture restores T3-mediated AKT phosphorylation
oxLDL impair T3-induced NO production
In light of the results obtained on AKT phosphorylation, we next evaluated the effects of LDL on T3-mediated NO production by using the NO-reactive dye DAF-2 DA, a specific and sensitive probe for a direct measurement of NO in single cells (21,22). A preliminary time course experiment showed that the fluorescent dye peaked after 20 minutes of T3 treatment (data not shown), while eNOS protein phosphorylation occurred after 60 minutes. This apparent discrepancy in response time most likely reflects a difference in sensitivity between Western blot and NO-specific fluorescent dye staining. Overnight-starved cells were incubated with oxLDL or nLDL before being exposed to T3 for 20 minutes. As expected, T3-stimulated NO production was significantly inhibited not only by pretreatment of endothelial cells with oxLDL (oxLDL+T3 vs. T3, 9.79±0.5 AU vs. 80.75±2.8 AU, p<0.0001), but also with nLDL pretreatment (nLDL+T3, 52.94±2.04 AU vs. 80.75±2.8 AU, p<0.0001), although pretreatment with nLDL, as compared with oxLDL, had a less pronounced effect on NO production (nLDL+T3 vs. oxLDL+T3, 52.94±2.04 AU vs. 9.79±0.5 AU, p<0.0001). NO production was restored by pretreatment with the antioxidants vitamin C and vitamin E (Fig. 3b, c). Taken together, these results strongly suggest that the oxidation of LDL is necessary for inhibiting T3-mediated NO production in endothelial cells.
oxLDL decrease T3-mediated NO-dependent cGMP production
NO activity is mediated by its target enzyme, cyclic guanylate cyclase, which converts GMP into cGMP. Immunoenzymatic analysis of cGMP was carried out to evaluate the bioavailability of NO by measuring the NO-dependent cGMP production to confirm the data obtained with DAF-2 DA fluorescence. The cells were treated using the same experimental conditions adopted for the DAF-2 DA assay. T3-stimulated cells, as compared with non-stimulated cells, had elevated cGMP levels (0.16±0.03 vs. 0.02±0.01 pmol/mL protein, respectively, p<0.0001); the increase in cGMP was attenuated by nLDL and drastically inhibited by the addition of oxLDL (Fig. 4). The presence of the antioxidant mixture of vitamins C and E prevented this phenomenon by restoring cGMP levels. These results strongly suggest that LDL oxidation is crucial in the inhibition of T3-mediated production of NO-dependent cGMP.
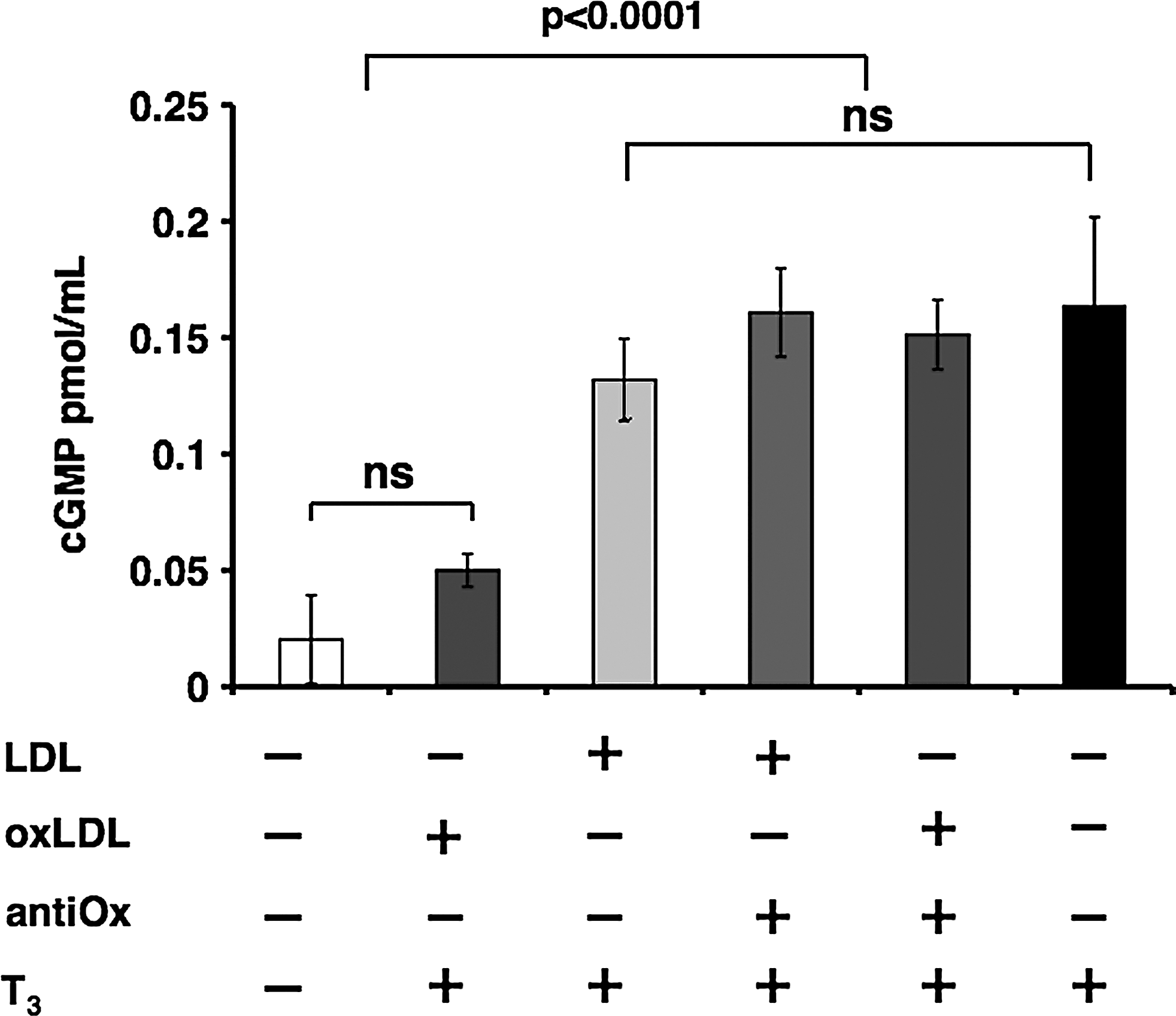
Effect of LDL oxidation on T3-mediated intracellular cyclic guanosine monophosphate (cGMP) production. Immunoenzymatic analysis of cGMP. HUVECs were treated for 20 minutes with T3 (10−7 M) or pretreated for 3 hours with oxLDL (50 μg/mL) or nLDL (50 μg/mL) and then stimulated with T3 (10−7 M). When indicated, cells were pretreated for 12 hours with the antioxidants. cGMP levels increased in T3-stimulated cells versus non-stimulated (p<0.0001); the increase in cGMP was attenuated by nLDL and drastically inhibited by oxLDL. The presence of the antioxidant mixture prevented this phenomenon by restoring cGMP levels. Data are reported as mean of three independent experiments±SD.
Discussion
The integrity of endothelial vascular function and the production of NO are essential for the regulation of vascular blood flow, and a dysfunction in this system may lead to hypertension and atherosclerosis (23,24). LDL oxidation is a key step in the atherogenesis process, and oxLDL may impair endothelium-dependent vessel relaxation (25,26). OxLDL adversely influence the endothelium by inducing the expression of adhesion molecules, stimulating apoptosis after long-term exposure, and impairing vasodilation (27,28). Experimental evidence suggests that oxLDL may alter various signaling pathways, such as PKC, MAPK, SAPK, tyrosine kinases, and the sphingomyelin/ceramide pathway, and transcription factors, namely AP1, NF-κB, and STATs, all of which are potentially involved in the atherosclerotic process (29 –31). oxLDL can also affect the PI3K/AKT pathway by interfering with insulin and VEGF signal transduction in human fibroblasts (32) and endothelial cells, respectively (33). By a mechanism not yet fully understood, oxLDL may also induce dephosphorylation of AKT at serine 473 and may impair the angiogenic properties of endothelial precursor cells by down-regulating E-selectin and integrin alpha(v)beta (5), resulting in a decrease in cell–cell interactions (33,34). While TH has a clear role in endothelial function, relatively little is known about the mechanism that regulates the interaction of T3 with endothelial cells, and to the best of our knowledge, no information is available on the effects of oxLDL on T3 action. This is of potential clinical relevance, as both endothelial dysfunction and LDL oxidation are common events in cardiovascular disease, and thyroid dysfunction is a frequent condition that confers an increased risk for cardiovascular complications.
In the present study, we provide evidence that oxLDL interfere with peripheral non-genomic T3 action by decreasing its ability to activate PI3K/AKT signaling, ultimately inhibiting NO production. We also observed this phenomenon when endothelial cells were treated with nLDL, but the effect was prevented by pretreatment with antioxidants, restoring the non-genomic activity of T3, the generation of NO, and cGMP production. We interpreted this result in light of the intracellular oxidative modification of nLDL that took place during the incubation of LDL which was necessary in our experimental conditions.
This is consistent with previous in vitro studies which showed that several cells, including macrophages, endothelial, and smooth muscle cells, may play a role in generating oxLDL from nLDL (35,36). For instance, Morel et al. showed that at least 2 hours of incubation of LDL with endothelial cells in serum-free medium, a condition similar to our experiments, was required to initiate the process that resulted in LDL modification from a native to an oxidized state (37). Furthermore, the authors showed that the presence of the antioxidant butylated hydroxytoluene prevented the modification of LDL (37). Another study proved that the incubation of LDL with smooth muscle cells resulted in LDL oxidation, which was prevented by superoxide dismutase, suggesting the key role of O2 − production by smooth muscle cells in LDL modification (38). Furthermore, our previous study demonstrated that platelets converted nLDL to oxLDL via a free radical-mediated mechanism involving GP91phox, as showed by enhanced conjugated dienes, lysophosphatidylcholine formation, electrophoretic mobility, Apo B-100 degradation, and increased LDL uptake by human macrophages (17). Taken together, these studies suggest that oxidation of nLDL might partially account for the nLDL effects on T3 action that were observed. These data are further supported by the protective effect of vitamin E and vitamin C that can prevent the oxidation of nLDL to oxLDL (39,40) and, consistent with our results, another study indicates that vitamin E partially restored insulin activated-signaling events, including AKT phosphorylation, impaired by pretreatment with oxLDL (32). However, the mechanisms whereby the antioxidants mixture may possibly revert the observed adverse effect of oxLDL on T3-mediated AKT phosphorylation is not clear, and further investigations are necessary to elucidate this issue. This study may have clinical relevance, as in hypothyroid patients the damaging effects of oxLDL on T3-mediated vasodilatation would be further amplified by the low circulating levels of T3 and by the intrinsic effects of hypothyroidism on serum lipid concentration (41). Thus, the combination of high levels of LDL and reduced T3-mediated vasodilation could further increase SVR and ultimately increase the risk for endothelial dysfunction, hypertension, and atherosclerosis. Therefore, our findings suggest a novel reciprocal relationship between T3 and LDL in the regulation of endothelial function.
In conclusion, we demonstrate that LDL oxidation may contribute to a blunting of the non-genomic T3 action on vascular endothelium, suggesting that oxLDL, apart from inducing the atherosclerotic process, may also promote a mechanism of peripheral pathway-specific resistance to T3.
Footnotes
Acknowledgments
This work was supported by Sapienza University of Rome and in part by the Intramural Research Program of the NIDDK-NIH, program Z01-DK047057-01 (F.S.C.). The authors would like to thank Dr. David Heber (UCLA Center for Human Nutrition, David Geffen School of Medicine, UCLA) for the critical revision of this article.
Disclosure Statement
No competing financial interests exist.