
Abstract
Background:
Cushing's syndrome (CS) due to ectopic adrenocorticotrophic hormone (ACTH) and/or ectopic corticotropic releasing hormone (CRH) secretion accounts for <10% of all CS. Neuroendocrine carcinomas rarely cause CS. These carcinomas have been found to secrete either ACTH or rarely CRH. Herein we report a case of neuroendocrine carcinoma originating from the thyroid as the source of ACTH-dependent CS.
Summary:
A 30-year-old woman with features of CS presented with severe respiratory distress. Six months before that, she was diagnosed with primary hypothyroidism and started on levothyroxine (LT4) therapy. Biochemical evaluation was done, and nonsuppressed serum cortisol levels following dexamethasone with high ACTH confirmed a diagnosis of ACTH-dependent CS. Magnetic resonance imaging of the brain showed a bulky pituitary gland. Adrenal imaging showed bilateral adrenal hyperplasia. A computerized tomography scan showed a large anterior mediastinal mass arising from the neck and extending behind the transverse aortic arch. She underwent emergency thoracotomy due to rapidly progressive superior mediastinal syndrome and left vocal cord palsy. At surgery, the mass was seen originating from the thyroid and the thymus was compressed posteriorly. Near total thyroidectomy and thymectomy with removal of pericardial seedlings were done. Histopathology revealed sheets, cords, and nests of round or oval tumor cells with hyperchromatic nuclei and scant cytoplasm with local invasion and lymphovascular embolization suggestive of a neuroendocrine carcinoma arising from thyroid, staining positive for cytokeratin, synaptophysin, and chromogranin-A, and negative for calcitonin and carcinoembryonic antigen.
Conclusions:
Here we report a case of a neuroendocrine tumor of the thyroid causing ACTH-dependent CS. The tumor was negative for calcitonin staining, indicating that this was not a medullary carcinoma of the thyroid. Neuroendocrine carcinomas originating from the thyroid gland are very rare. A thyroid tumor of neuroendocrine origin causing ACTH-dependent CS has not been reported previously.
Introduction
Neuroendocrine carcinoma is an extremely rare variant of a heterogeneous group of epithelial tumors, and has been reported as a cause of ectopic CS (5–6). Nonmedullary cell neuroendocrine carcinomas arising from the thyroid gland account for <1% of all neuroendocrine tumors (7), but these neuroendocrine tumors of the thyroid have not been previously reported to cause ectopic CS. Here we report a case of a neuroendocrine tumor of the thyroid (not medullary carcinoma) which, putatively, was producing a substance which is simulating ACTH production, leading to excess production of cortisol.
Patient
A 30-year-old woman presented with a history of rapid weight gain, fatigability, proximal muscle weakness, easy bruising, acne, hirsuitism, facial swelling, increased skin pigmentation, and recurrent skin infections for 5 months, amenorrhea for 2 months, and episodes of palpitation, diaphoresis, dry mouth, and respiratory distress for a month before presentation (Fig. 1). Clinical examination revealed tachycardia (∼140 beats/minute), tachypnea (∼22 breaths/minute), hypertension (180/110 mmHg with significant postural drop), diaphoresis, and typical features of CS. She had a small soft diffuse thyroid, which was palpable and was visible only on neck extension. The Pemberton's sign was positive.
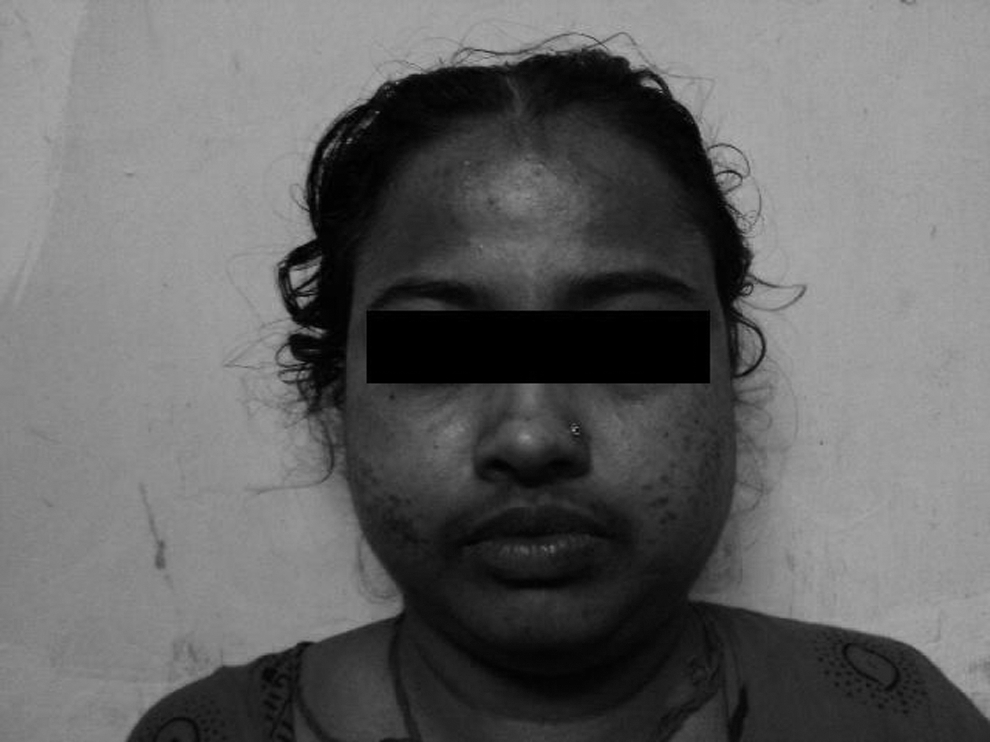
Moon face, hirsuitism, and acne typical of Cushing's syndrome.
She had been diagnosed with primary hypothyroidism 6 months previously, having an initial serum-free thyroxine of 0.84 ng/dL and a serum thyrotropin (TSH) of 22 mIU/L. She was taking 75 μg levothyroxine (LT4) daily. At admission, she had severe hypokalemia (serum K=1.8 mmol/L) with alkalosis. Subsequently, the serum potassium fluctuated between 2.2 and 3.1 mmol/L despite potassium infusion and oral potassium supplementation. Her serum cortisol was 28 μg/dL, not suppressed, despite having taking 8 mg dexamethasone the previous night. After taking 1 mg of dexamethasone every 6 hours for 2 days, the serum cortisol was still not suppressed and was 22.2 μg/dL. After taking 2 mg of dexamethasone every 6 hours for 2 days, the serum cortisol was 31 μg/dL. ACTH was not suppressed and was 124 pg/mL (Table 1). The repeat serum TSH along with serum luteinizing hormone, follicle-stimulating hormone, prolactin, fasting blood glucose, and 24-hour urine fractionated metanephrines were all normal. Magnetic resonance imaging (MRI) of the brain (with contrast) revealed a bulky pituitary (13 mm×9 mm×7 mm) with homogenous contrast uptake (Fig. 2). We were able to measure ACTH levels in the patient preoperatively. Basal 8 AM cortisol was done using a chemiluminescence assay (Immulite 1000, Siemens, Gwynedd, United Kingdom). CRH assay was not available at our institute and, hence, could not be performed. A computerized tomography (CT) of the abdomen showed bilateral adrenal hyperplasia (Supplementary Fig. S1; Supplementary Data are available online at
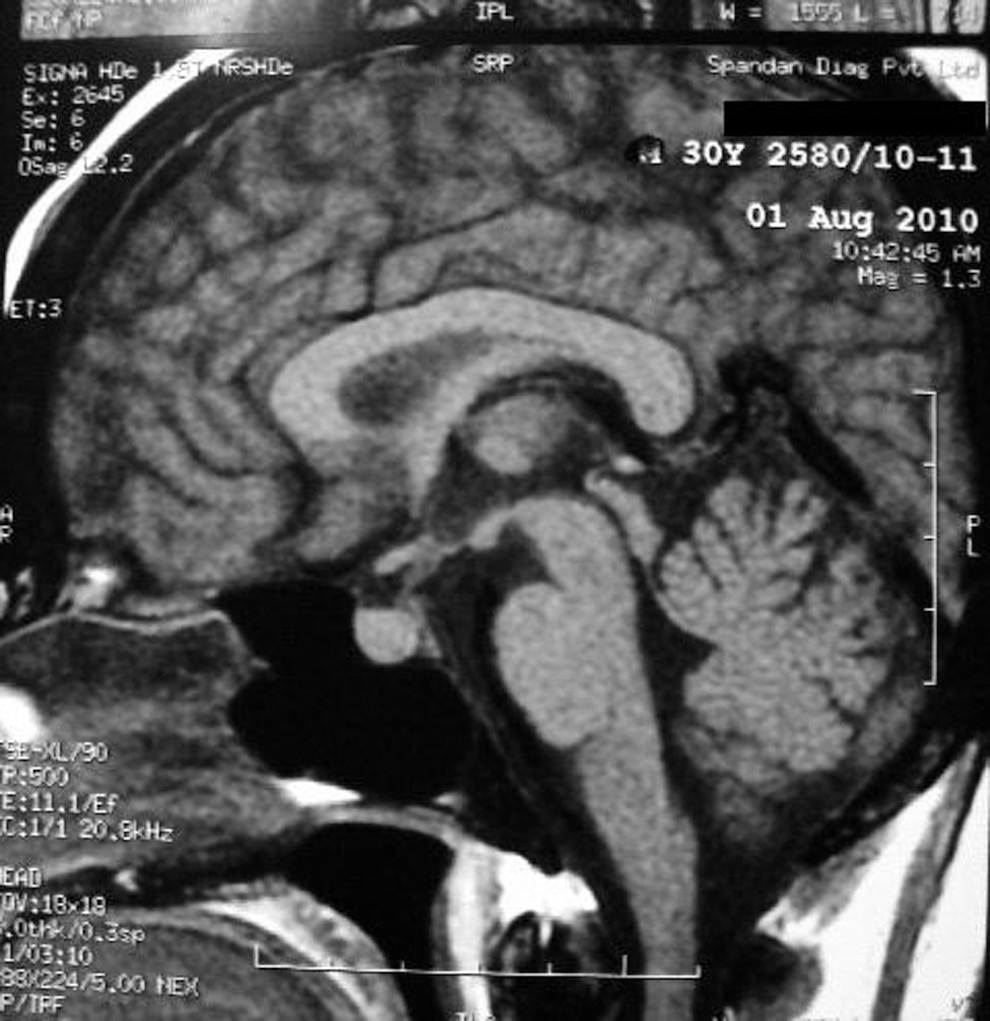
T1 weighted image of pituitary showing pituitary hyperplasia.
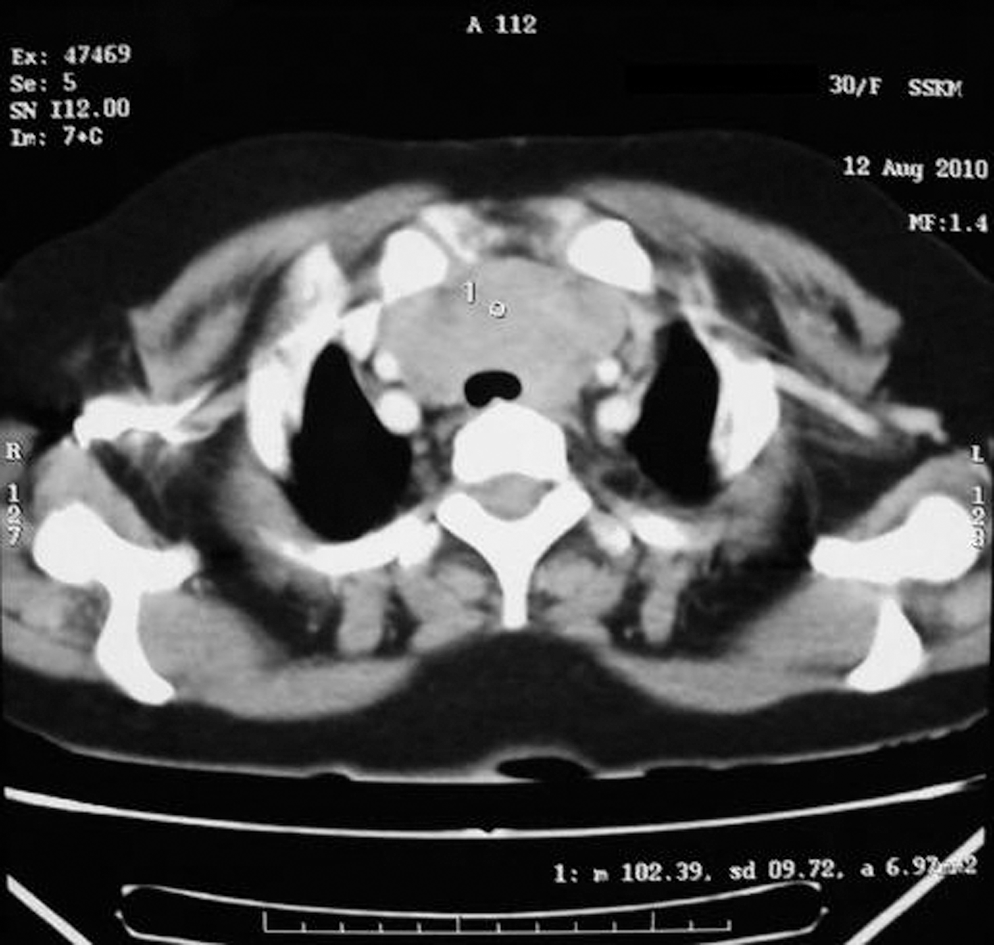
Section of the upper thorax showing an anterior mediastinal mass.
ACTH, adrenocorticotrophic hormone; HDDST, high-dose dexamethasone suppression test; ONDST, overnight dexamethasone suppression test.
She underwent emergency thoracotomy. An anterior mediastinal mass arising from the left lower pole of the thyroid, suggestive of a retrosternal thyroid mass, was found compressing the thymus posteriorly. Near total thyroidectomy with thymectomy was done with the removal of pericardial masses of ∼5–10 mm. The mass was encasing the major vessels and nerves in the thoracic inlet, necessitating removal of a part of both recurrent laryngeal nerves and a part of the innominate vein. The mass was encasing the trachea and, hence, the patient needed a preoperative tracheostomy.
She developed hypocalcaemia on postoperative day 2, which was managed with calcium infusions. Postoperatively there was normalization of her blood pressure and serum cortisol levels (Table 1). Histopathology of the mass revealed sheets, nests, and cords of small tumor cells with round or oval hyperchromatic nuclei and scanty cytoplasm arising from the thyroid (Fig. 4). The tumor had invaded the fibroadipose tissue surrounding the thyroid and had infiltrated the thymus (Supplementary Fig. S3). There was evidence of lymphovascular invasion (Supplementary Fig. S4). The nodules over the pericardium had features of the main tumor and were considered to be metastases from that tumor. The tumor cells stained positive for cytokeratin, synaptophysin, chromogranin-A, and were immunonegative for calcitonin and carcinoembryonic antigen (CEA; Supplementary Figs. S5–S9). Hence, a diagnosis of a nonmedullary neuroendocrine tumor of the thyroid was made.
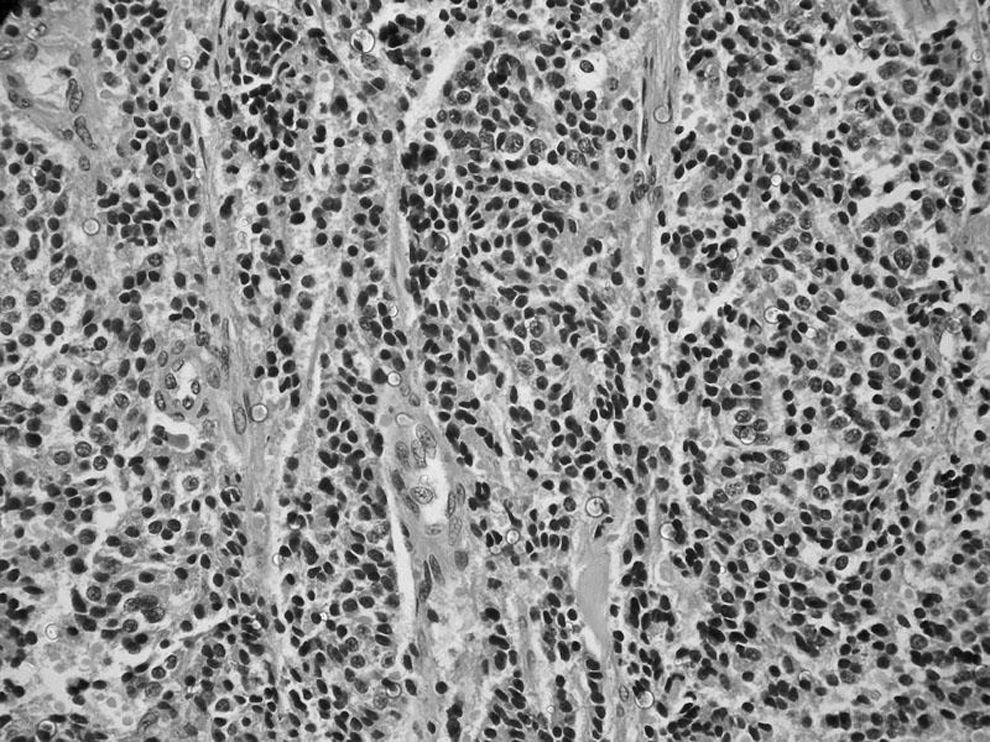
Sheets, cords, and nests of small round and oval tumor cells with hyperchromatic nuclei.
Discussion
Neuroendocrine tumors of the thyroid are an extremely rare group of tumors and include C-cell lesions, mixed C-cell– and follicular-derived tumors, paragangliomas, intrathyroidal parathyroid adenomas, secondary neuroendocrine tumors of the thyroid, and neuroendocrine carcinoma, with thyroid paragangliomas being the most common type (8). C-cell tumors, namely, medullary carcinoma of thyroid and C-cell hyperplasia and mixed medullary tumors produce calcitonin and stain positive for calcitonin. Medullary carcinomas of the thyroid can rarely produce ACTH and cause CS (9). However, the absence of immunoreactivity for calcitonin and CEA ruled out this entity in this patient. Few reports of neuroendocrine tumor metastases to the thyroid from gastropancreatic neuroendocrine tumors have been reported (10,11). Thymomas can be easily differentiated from neuroendocrine tumors on histochemistry and by the absence of neuroendocrine markers. Thyroid paragangliomas are rare. To date, only 25 such cases have been reported in the literature, and none of them have been reported to cause CS. These tumors usually present in women with solitary thyroid masses with typical round cell arranged in nests. Paragangliomas lack significant cellular atypia, mitotic activity, necrosis, vascular invasion, and cytokeratin expression, which are consistently seen in neuroendocrine carcinomas (12,13). Neuroendocrine carcinomas classically stain positive for cytokeratin, synaptophysin, and chromogranin-A and show evidence of vascular invasion.
The tumor cells in our patient stained positive for cytokeratin, synaptophysin, chromogranin-A, and were immunonegative for calcitonin and CEA. Hence, a diagnosis of a nonmedullary neuroendocrine carcinoma of the thyroid was made.
Neuroendocrine carcinomas are an extremely rare variant of neuroendocrine tumors. There has been a single case report of a neuroendocrine carcinoma of the mesentery causing CS (7). Moreover, there have been few reports of neuroendocrine carcinomas of the thymus causing CS (8). A neuroendocrine carcinoma originating from the thyroid causing ACTH-dependent CS has not been reported as of yet.
Ectopic CS is caused by inappropriately raised levels of ACTH, either due to direct tumor production of ACTH or due to the secondary increase in ACTH because of the rise of corticotrophin-releasing hormone (CRH), which in turn stimulates the pituitary gland to increase ACTH secretion. Tumors causing ectopic CS may secrete either true ACTH or CRH, or molecules similar to them. Fifteen percent of cases of CS are associated with nonpituitary secreting ACTH (14). Ectopic CRH secretion causing CS is a very rare cause of ectopic ACTH-dependent CS, accounting for <1% of all CS (15).
A CRH assay and CRH staining facilities were not available. Our patient clearly had inappropriately raised ACTH levels and nonsuppressed cortisol levels, and typical clinical features of CS. This is consistent with a diagnosis of ACTH-dependent CS. The fact that the patient had bilaterally enlarged adrenals and a bulky pituitary on MRI, makes it likely that the thyroid tumor was producing a substance, such as CRH or a CRH-like substance.
The course of the disease was rapidly progressive and there was evidence of compressive symptoms that necessitated emergency thoracotomy with tracheostomy for decompression and removal of the anterior mediastinal mass. Normalization of the postoperative cortisol levels confirmed that the mass was the lesion responsible for the CS. This case also highlights the fact that a retrosternal thyroid mass should be considered in the differential diagnosis of anterior mediastinal masses. A positive ACTH or CRH immunoreactivity in the mass that was removed would have been of further help in confirming the diagnosis, but the postoperative clinical and biochemical parameters are consistent with an effective treatment of the CS and suggest that the resected mass was the source of the stimulatory factor causing CS. To the best of our knowledge, this is the first reported case of a neuroendocrine carcinoma originating from the thyroid causing ACTH-dependent CS.
Footnotes
Author Disclosure Statement
No competing financial interests exist.