
Abstract
Background:
The aryl hydrocarbon receptor (AhR) is a transcription factor that is activated by xenobiotic substances such as dioxin. After activation, it binds to dioxin response elements of DNA, thereby inducing transcription of a variety of xenobiotic metabolizing enzymes. To investigate whether AhR-activating substances accumulate in patients with endocrine disorders, we tested serum samples for AhR-stimulating activity.
Methods:
Serum AhR-stimulating activity was evaluated by exposing the HepG2 cells transiently transfected with an AhR-responsive reporter plasmid to serum samples. On the basis of preliminary findings that implicated methimazole (MMI), wild-type and AhR-null mice were treated with MMI, and their plasma AhR-stimulating activities and thyroxine levels were quantified.
Results:
In 28 randomly chosen patients, 7 out of 10 Graves' disease patients exhibited increased serum AhR-stimulating activity. The increased activity did not correlate with thyroid hormone status. However, we hypothesized that it might be caused by MMI. Subsequent analyses revealed that in 25 of 26 MMI-treated Graves' patients, serum samples collected after the MMI treatment had significantly higher AhR-stimulating activity compared to samples obtained when the same patients were not on MMI. By contrast, serum AhR-stimulating activity was unchanged in samples from the seven patients on propylthiouracil (PTU) compared to serum taken before the PTU treatment. In vitro experiments demonstrated that an MMI metabolite 3-methyl-2-thiohydantoin, but not MMI, activated AhR. MMI increased plasma AhR-stimulating activities and reduced plasma thyroxine concentrations, in both wild-type and AhR-deficient mice.
Conclusions:
Graves' patients taking MMI have increased serum AhR-stimulating activity, which is unrelated to thyroid hormone status, but correlates with MMI treatment. The AhR activation is likely caused by 3-methyl-2-thiohydantoin. Further studies are required to determine the potency of 3-methyl-2-thiohydantoin as an AhR activator and the significance of the differences between MMI and PTU observed in this study.
Introduction
In this study, we examined the serum of patients with various endocrine disorders for AhR-stimulating activity using a sensitive reporter gene assay. Initial studies revealed high levels of AhR-stimulating activity in Graves' disease patients taking methimazole (MMI), prompting us to test whether MMI is responsible for this activity.
Materials and Methods
Chemicals
The following chemicals were purchased: β-naphthoflavone (β-NF; Sigma-Aldrich, St. Louis, MO), AhR antagonist CH-223191 (Calbiochem, San Diego, CA), 1-methyl-2-mercaptoimidazole (MMI; Sigma-Aldrich), propylthiouracil (PTU; Sigma-Aldrich), L-thyroxine sodium salt pentahydrate (Sigma-Aldrich), 3-methyl-2-thiohydantoin (MTH-Glycine, Research Organics, Cleveland, OH), and 1-methylimidazole (Sigma-Aldrich).
Subjects
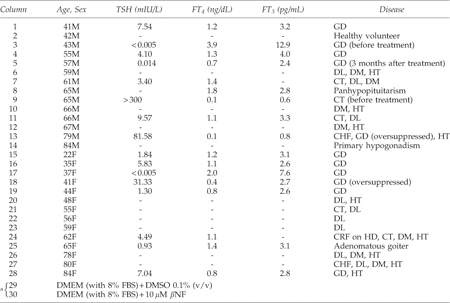
This study was approved by the ethics committee of our institution (reference number: 07–10). All the subjects, who were seen at the Teikyo University Hospital from September 1, 2006, through March 31, 2009, gave their written consent for their serum samples obtained in routine clinical practice to be analyzed. Aliquots of serum samples were stored at −80°C until experiments were performed. The age given in each figure is the age when the patient was first seen in our hospital. All the patients treated with either MMI or PTU were patients with Graves' disease. We did not encounter any patient with toxic nodular goiter during the above-mentioned period, probably because of the extremely low prevalence of hyperfunctioning thyroid nodules in Japan.
Animals
Two pairs of heterozygous AhR-deficient mice (10), which had been backcrossed onto the DBA/2 background, were provided by the RIKEN BioResource Center (Tsukuba, Japan), and their offspring were used in this study. We chose to use male mice for our experiments, because the AhR activity could be affected by estrogen levels (11), which change depending on the phase of the estrous cycle in female mice. Twenty-four-day-old male mice were treated with MMI or PTU [0.1% (w/v) in drinking water] for 21 days, and sacrificed for plasma collection. In some wild-type mice, L-thyroxine sodium pentahydrate (1 mg/L in drinking water) was given along with MMI. Plasma samples were subjected to either the reporter gene assays or determination of thyroxine concentrations, which was carried out at the Nagahama Life Science Laboratory (Shiga, Japan). The experiments were approved by the animal ethics committee of our institution (reference number: 0804068A1b).
Plasmids
The AhR-responsive firefly luciferase-expressing plasmid DRE4-GL3 was constructed by inserting four typical DREs (5′-TGAGAATGTGCGTGACAAGGTCT-3′×4) (12) immediately before the SV40 promoter that drives transcription of the firefly luciferase gene. The parental pGL3-Promoter vector (abbreviated as GL3; Promega, Madison, WI) containing no DRE sequence was used in the control experiments. A Renilla luciferase plasmid phRL-CMV (Promega) was used as an internal control in the transfection experiments.
Reporter gene assays
HepG2 and MCF-7 cells, both of which express AhR and Arnt (13), were maintained in the Dulbecco's modified Eagle's medium supplemented with 8% (v/v) heat-inactivated fetal bovine serum and antibiotics. One day before transfection, the medium was replaced with a fresh medium. On the day of transfection, one 10-cm dish of cells (∼80% confluent) were trypsinized and split into one 96-well B&W IsoPlate (PerkinElmer, Waltham, MA), and 0.25 μg of either DRE4-GL3 or GL3 was tranfected, along with 0.1 ng of phRL-CMV, into each well, using the transfection reagent DreamFect (OZ Biosciences, Marseille, France). Four hours later, the medium containing transfection complex was replaced with 50 μL of the fresh medium containing the substance of interest, patient serum, or mouse plasma. For experiments using a medium, three wells were designated to one treatment condition. As for patient serum and mouse plasma samples, three wells were assigned to one serum sample taken at a given time from one patient, and one well was allocated to one plasma sample obtained from each of three mice that had been treated similarly. Twenty hours later, cells were lysed and measured for firefly and Renilla luciferase activities using the Dual-Luciferase Assay System (Promega), and normalized firefly luciferase activity (ratio of firefly/Renilla luciferase luminescence) was calculated. A series of serum samples from the same patient were analyzed simultaneously and their luciferase activities were shown in one graph.
Statistical analysis
Statistical analysis was done using analysis of variance with the Tukey-Kramer post hoc test, using StatView 5.0 software (SAS Institute, Cary, NC).
Results
In a pilot experiment, we analyzed the serum samples from 14 male and 14 female subjects who were randomly chosen from those seen in our endocrinology clinic (Fig. 1 and Table 1). HepG2 cells transiently transfected with either DRE4-GL3 or the parental GL3 plasmid were cultured directly in these serum samples for 20 hours, and were measured for standardized firefly luciferase activity. Sera from several patients exhibited strikingly (Fig. 1 and Table 1, columns 13 and 15) or mildly (Fig. 1 and Table 1, columns 4, 5, 17, 18 and 19) higher AhR-stimulating activity, regardless of the patients' age, gender, smoking status, the timing (fasting or postprandial, morning or afternoon) of blood collection, serum lipid profile, or plasma glucose level (data not shown). The only common feature of these patients was that they were Graves' disease patients on MMI. Because their thyroid hormone status (hyper-, eu-, or hypothyroidism) was unrelated to the serum AhR-stimulating activity, we hypothesized that MMI, but not abnormal thyroid function, induced AhR-stimulating activity. When MCF-7 cells were used instead of HepG2, similar results were obtained (data not shown).
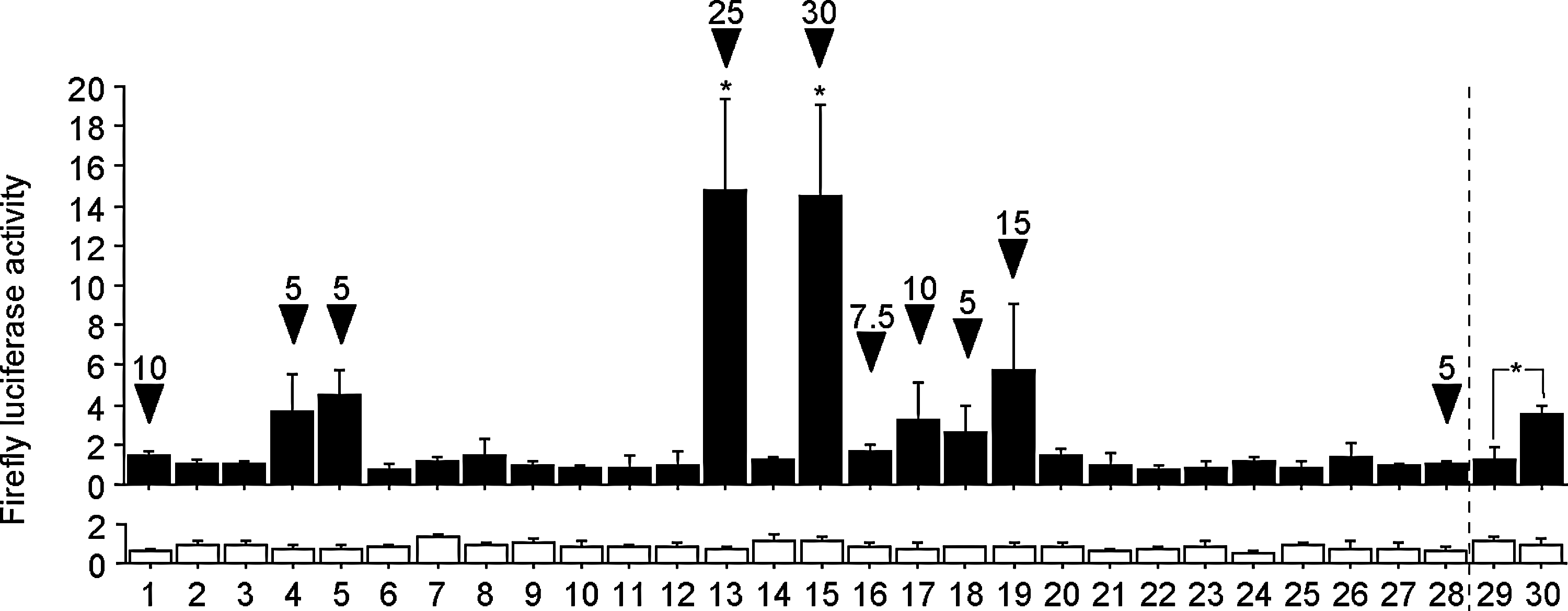
Serum AhR-stimulating activity in patients with different endocrine or metabolic diseases. Solid columns (upper graph) and open columns (lower graph) show the mean standardized-firefly luciferase activity in HepG2 cells transiently transfected with DRE4-GL3 and GL3, respectively. Each serum sample was tested in triplicate, and error bars represent SDs for the triplicate transfections. Profiles of the patients are shown below. The mean values in column 2, which represents a healthy subject, were defined as 1.0. Arrowheads denote MMI-taking patients, and the numbers above them represent daily MMI doses (mg/day). The validity of the assay was confirmed by demonstrating induction of DRE-mediated transcription by a known AhR agonist β-NF in columns 29 and 30, in which cells were cultured in the fetal bovine serum-supplemented Dulbecco's modified Eagle's medium with 0.1% (v/v) DMSO vehicle and 10 μM β-NF, respectively. Normal ranges for thyroid function tests are: TSH 0.34–4.50 mIU/L; FT4 0.8–1.8 ng/dL; FT3 2.0–4.5 pg/mL. As for the two patients who turned out hypothyroid unexpectedly, one (column 13) was severely hypothyroid, presumably because of his improved medication compliance. The other (column 18) was in the process of optimal dose determination. *Asterisks indicate significantly (p<0.05) higher luciferase activity than column 2. AhR, aryl hydrocarbon receptor; DRE, dioxin-response element; MMI, methimazole; β-NF, β-naphthoflavone; TSH, thyrotropin; FT4, free thyroxine; FT3, free triiodothyronine; DMSO, dimethyl sulfoxide.
HepG2 cells were cultured in DMEM with or without 10 μM βNF, to show the validity of this assay.
GD, Graves' disease; DL, dyslipidemia; DM, diabetes mellitus; HT, hypertension; CT, chronic thyroiditis; CHF, congestive heart failure; CRF on HD, chronic renal failure on hemodialysis.
We next analyzed samples from another two Graves' patients. Two serum samples were obtained from each patient, one during MMI therapy and the other without MMI treatment. Consistently with the preliminary observations in Figure 1, serum AhR-stimulating activity was higher when the patients were on MMI than when they were not (Fig. 2). The higher fold induction of AhR-stimulating activity by MMI in Figure 2B than in Figure 2A may be ascribed to the different MMI doses the patients were taking (5 and 30 mg/day in Fig. 2A and B, respectively), but other factors such as sex hormones might be involved in the difference of serum AhR-stimulating activity. In both Figure 2A and B, the elevated firefly luciferase activity was clearly reduced by addition of a potent and specific AhR antagonist CH-223191 (14), verifying that the higher luciferase activity resulted from activation of AhR.
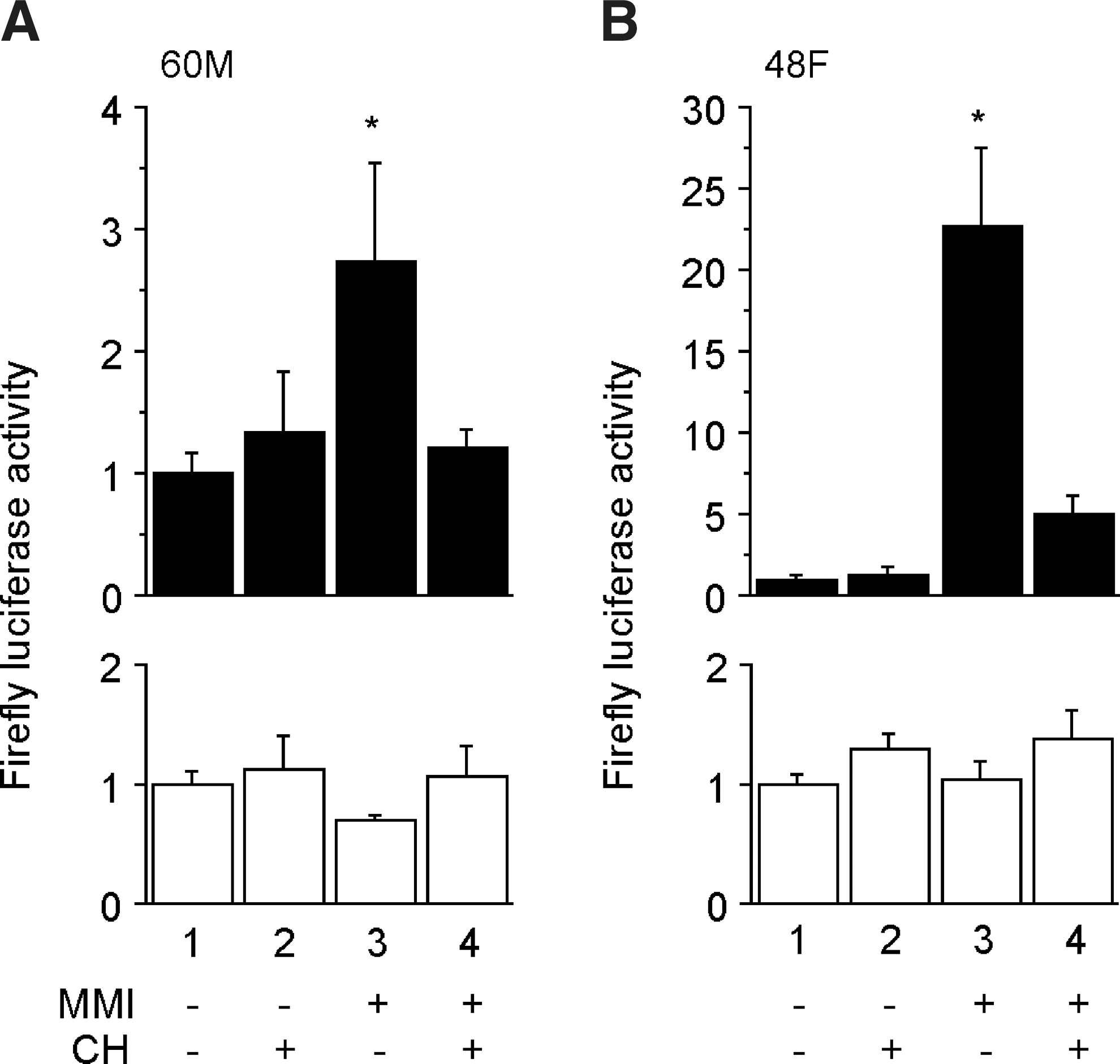
Effects of CH-223191 on MMI-induced serum AhR-stimulating activity.
We further investigated temporal changes in serum AhR-stimulating activity in the patients following initiation of treatment with MMI. First, we ascertained that serum AhR-stimulating activity did not fluctuate significantly over a long period, in a patient who did not take MMI (Fig. 3). In MMI-taking patients, MMI induced higher serum AhR-stimulating activity in some patients than in others (Fig. 4 and Supplementary Figs. S1 and S2; Supplementary Data are available online at
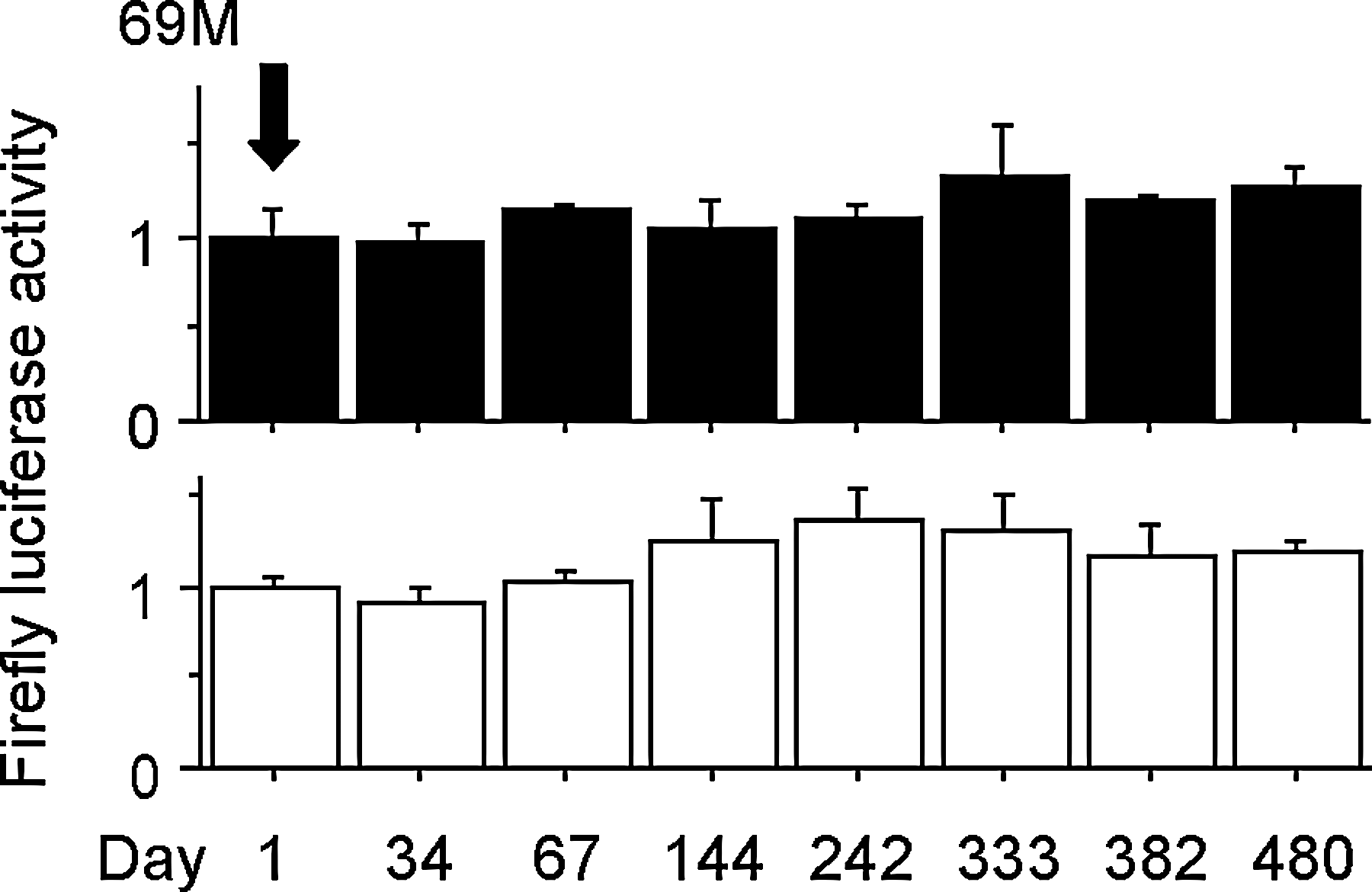
Temporal changes of serum AhR-stimulating activity in a 69-year-old patient with chronic thyroiditis. This patient was taking LT4. Solid columns (upper graph) and open columns (lower graph) show the mean standardized-firefly luciferase activity in HepG2 cells transfected with DRE4-GL3 and GL3, respectively. Error bars represent SDs for triplicate transfections. Arrow points to the standard column, whose mean value was defined as 1.0. Day 1 is defined as the day when the first serum sample was collected. SD, standard deviation; LT4, levothyroxine.
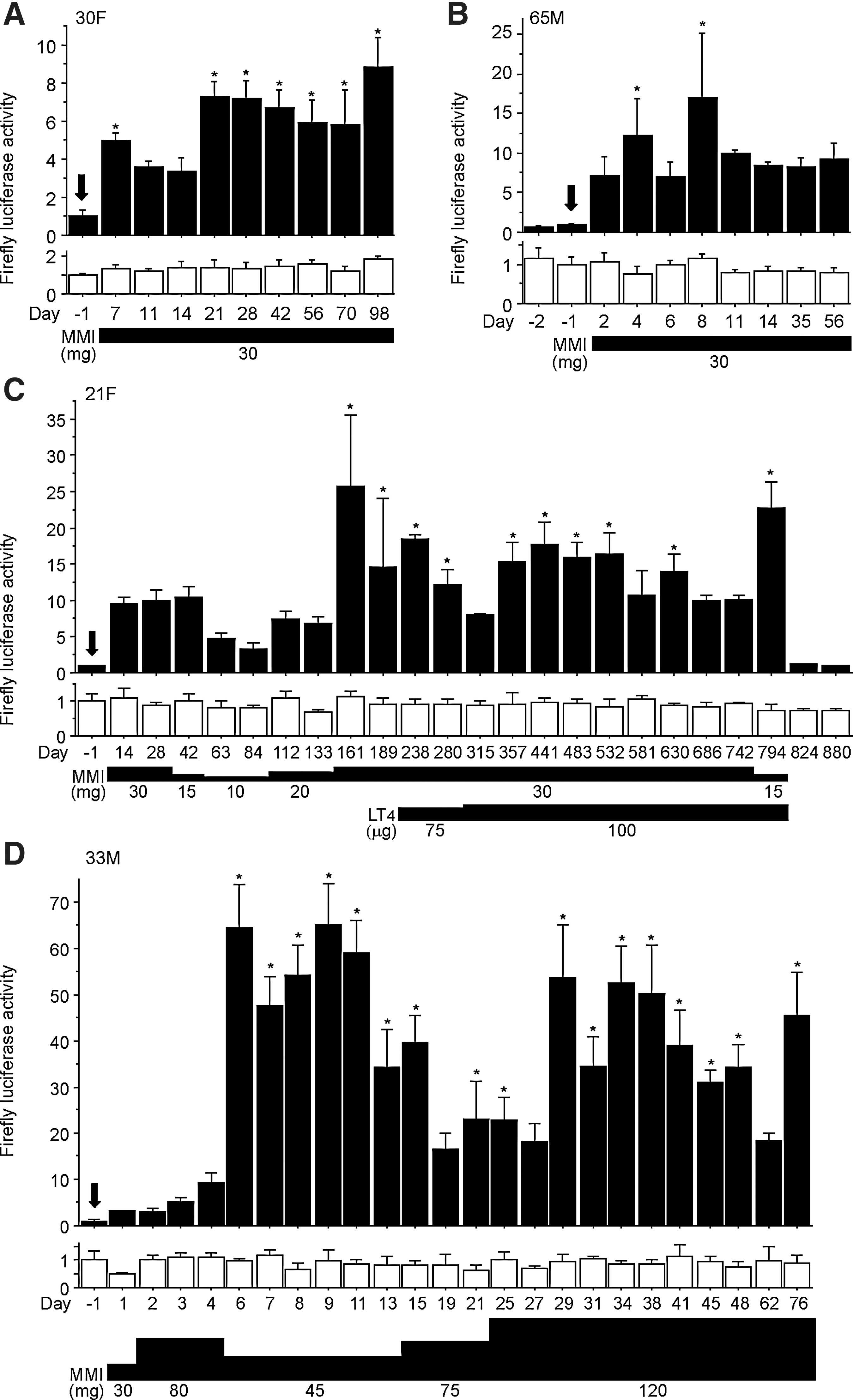
Representative data of temporal changes of serum AhR-stimulating activity in MMI-treated Graves' patients. Patients
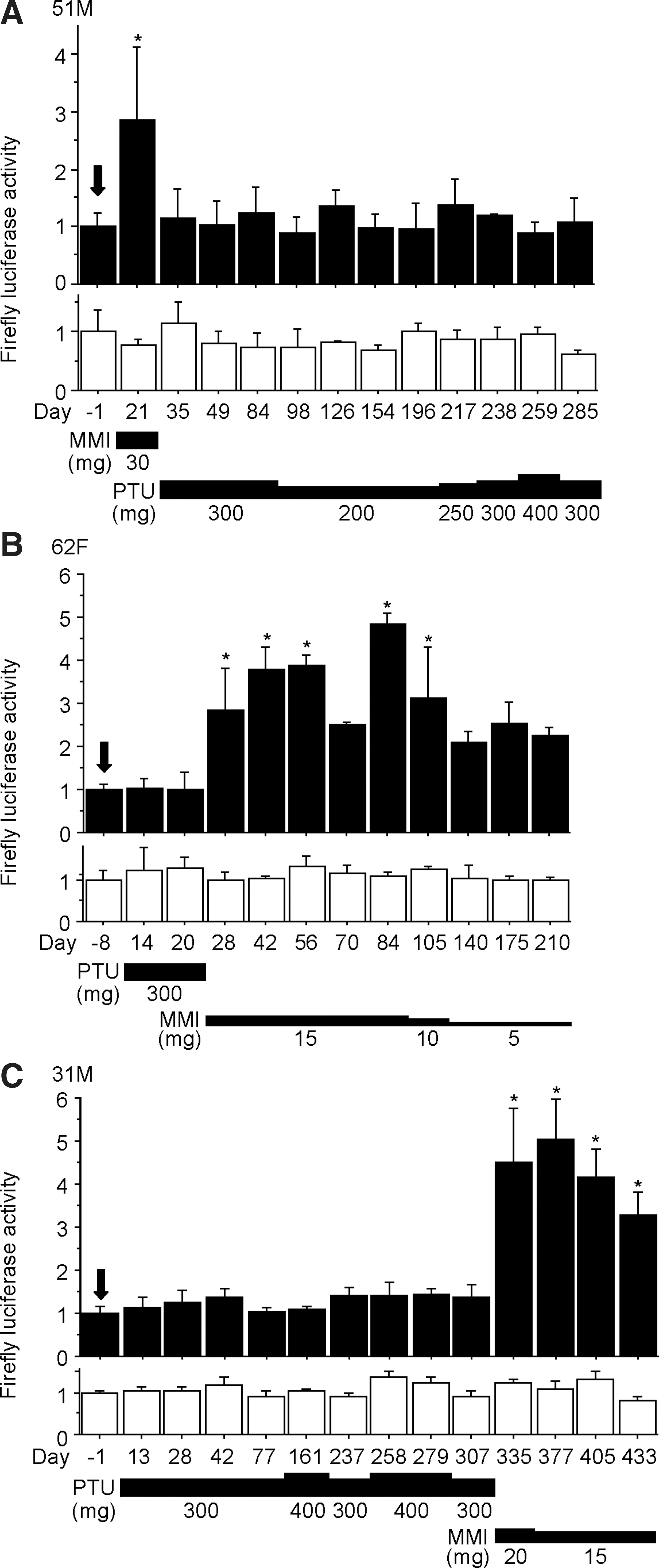
Temporal changes of serum AhR-stimulating activity in Graves' patients in whom one antithyroid drug was switched to the other. Antithyroid drugs were switched because the initial drug caused adverse side effects [pruritus and liver injury in patients
The effects of another antithyroid drug, PTU, were also investigated. Among the 7 patients taking PTU (Fig. 5 and Supplementary Fig. S3), none exhibited enhanced serum AhR-stimulating activity.
We then tried to identify the substance(s) that activated AhR in MMI-treated patients. Given the relatively rapid appearance of the serum AhR-stimulating activity (1 day or 2 after MMI administration [e.g., Fig. 4B, D]), it was reasonable to assume that MMI itself or its immediate metabolite(s) acted as AhR agonist(s). Therefore, we tested whether MMI and its two main metabolites, 3-methyl-2-thiohydantoin and 1-methylimidazole (15), could stimulate AhR. As shown in Figure 6A, 3-methyl-2-thiohydantoin added to the culture medium clearly enhanced AhR-mediated transcription dose dependently, while MMI and 1-methylimidazole as well as PTU did not. Because it has been reported that serum 3-methyl-2-thiohydantoin concentration after a single oral administration of 60 mg MMI became detectable (∼0.1 μg/mL) at 2 hours, reached 0.3 μg/mL at 12 hours, and then gradually declined to ∼0.15 μg/mL at 24 hours in one hyperthyroid patient (16), we tested whether this range of concentrations of 3-methyl-2-thiohydantoin could activate AhR, in the absence (Fig. 6A) or presence (Fig. 6B) of human serum. In the culture medium, 3-methyl-2-thiohydantoin stimulated AhR activity at concentrations of 1μg/mL and above (Fig. 6A). In serum, a lower concentration (0.1 μg/mL) of 3-methyl-2-thiohydantoin robustly stimulated AhR-mediated transcription (Fig. 6B), indicating that this substance is an AhR activator at concentrations encountered in a treated patient. Thus, 3-methyl-2-thiohydantoin may be responsible for serum AhR-stimulating activity that was observed after MMI administration, although other unknown MMI metabolite(s) might also be involved in AhR activation.
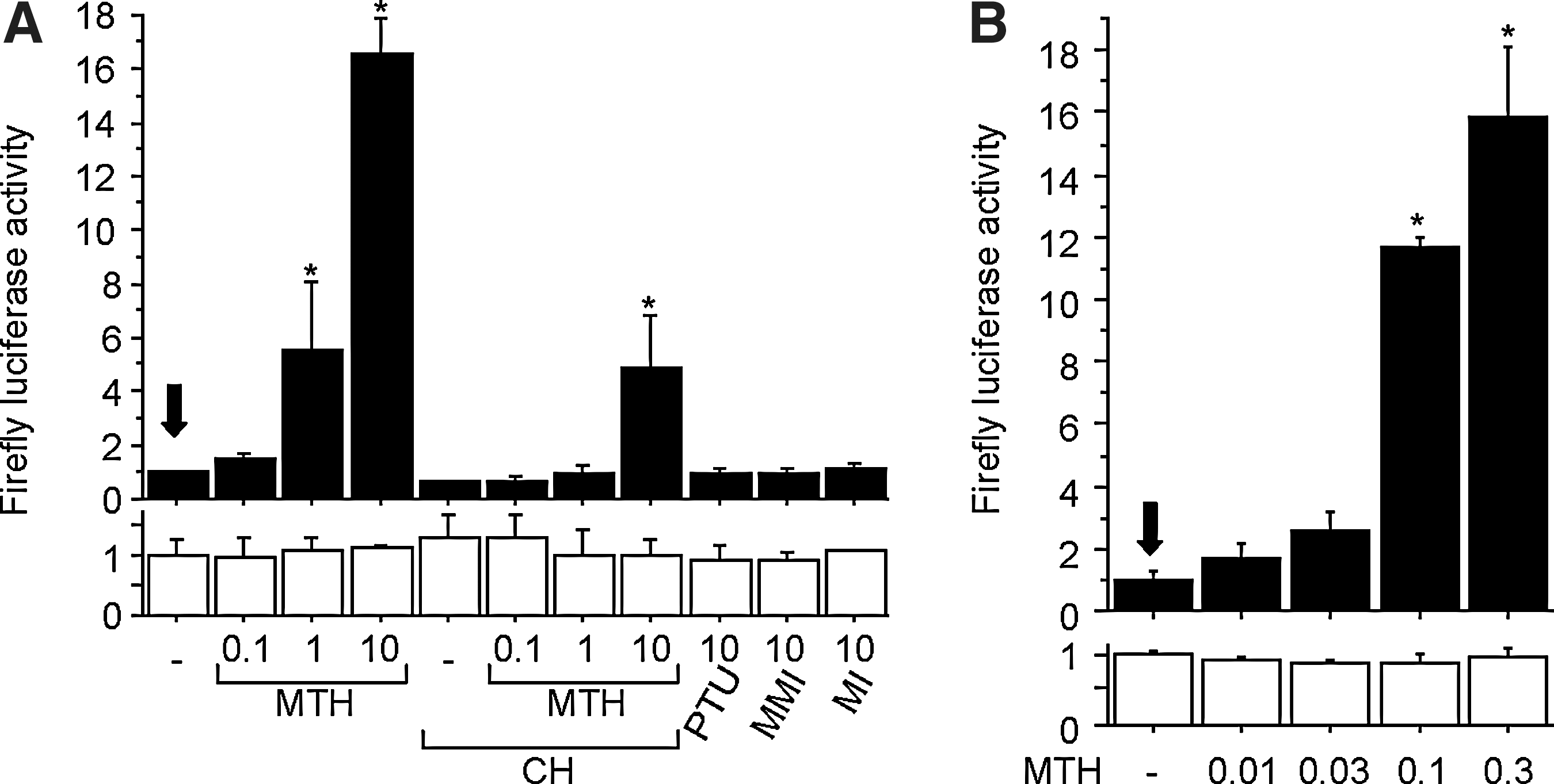
Activation of AhR by 3-methyl-2-thiohydantoin.
We then tested whether the effects of MMI administration on serum AhR-stimulating activity could be reproduced in mice. To get further insights, we used not only wild-type, but also AhR-null mice. Plasma samples obtained from wild-type, hetero- and homozygous AhR-deficient male mice treated with MMI, but not PTU, showed elevated AhR-stimulating activity (Fig. 7A), suggesting that comparable amounts of AhR agonist(s) accumulated in wild-type and AhR-deficient mice after MMI treatment. In both wild-type and AhR-null mice, plasma thyroxine concentrations clearly declined after 3-week-administration of either MMI or PTU (Fig. 7B). As was already suggested in Figure 4, hypothyroidism caused by MMI was unrelated to plasma AhR-stimulating activity. In addition, AhR activation by plasma was observed in the wild-type mice whose plasma thyroxine levels were maintained normal by administering both MMI and thyroxine concurrently (Fig. 7). Taken together, it can be concluded that MMI induces AhR-stimulating activity in mice as well regardless of thyroid hormone status, and that MMI suppresses thyroid function even without activation of AhR.
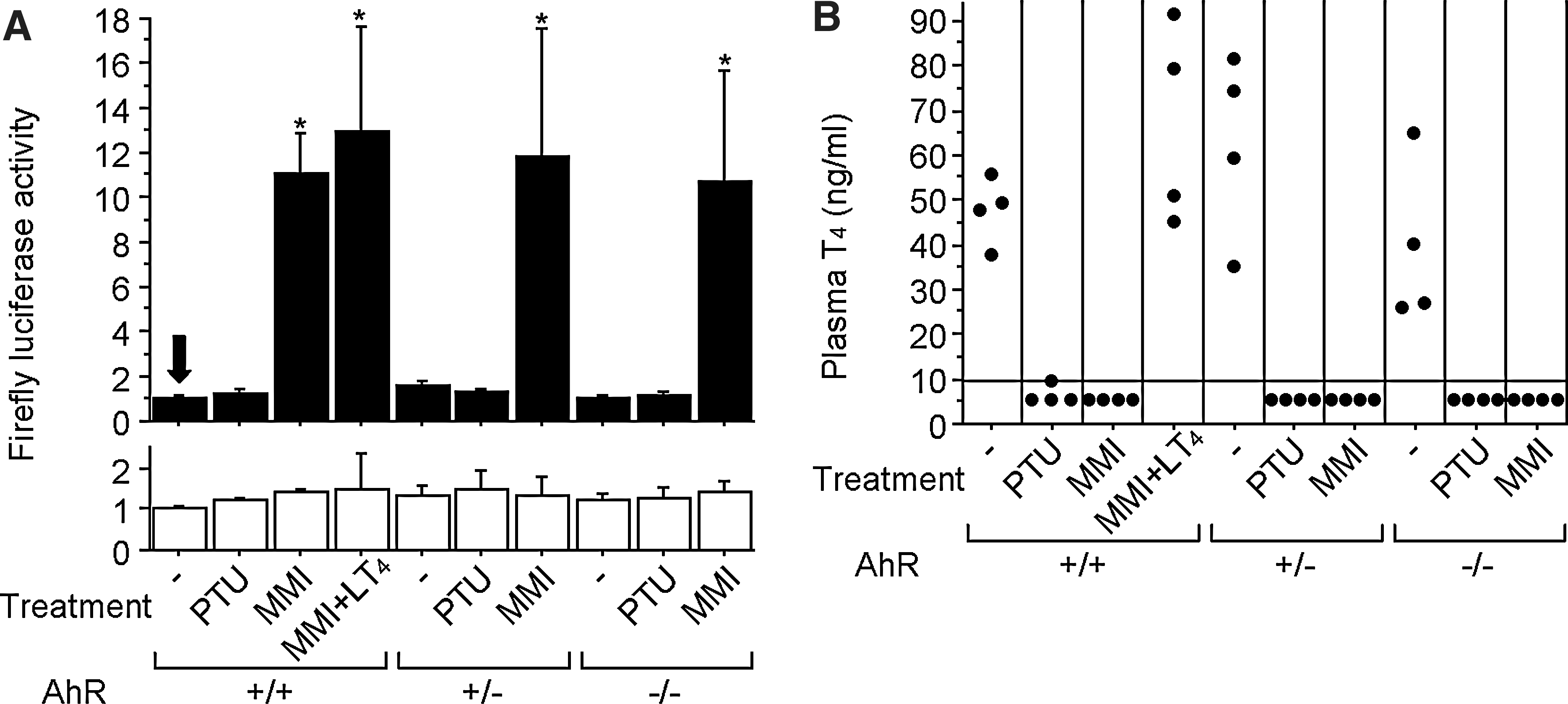
Effects of antithyroid drugs on wild-type and AhR-deficient mice.
Discussion
We started our study by testing serum AhR-stimulating activity in various endocrine patients, and suspected that the MMI treatment might be linked to higher serum AhR-stimulating activity regardless of thyroid hormone levels. In the following experiments, we confirmed that administration of MMI, but not PTU, induced activation of AhR, which was clearly blocked by adding an AhR antagonist CH-223191 to serum samples. Furthermore, treatment with MMI, but not PTU, enhanced plasma AhR-stimulating activity in mice as well. The AhR stimulation seemed to be ascribed to an MMI metabolite 3-methyl-2-thiohydantoin, but not MMI itself. It has been reported in a previous study that 3-methyl-2-thiohydantoin was not detected in all patients' serum samples after treatment with a single dose of 60 mg of MMI (16), indicative of variation in its serum concentrations among patients. This might account for why serum AhR-stimulating activity differed considerably among patients and was not elevated in a few MMI-treated patients in our study. To clarify it, we are now trying to establish a sensitive and reliable method to measure 3-methyl-2-thiohydantoin levels in our patients' serum samples.
Activation of AhR induces transcription of many different target genes (17,18). First, AhR activation upregulates the phase I cytochrome P450 enzymes, including CYP1A1, CYP1A2, and CYP1B1, which play a pivotal role in metabolizing xenobiotics. Second, activated AhR enhances expression of the phase II-conjugating enzymes, such as uridine diphosphate glucuronosyltransferase (UGT) 1A6, NAD(P)H-dependent quinone oxydoreductase-1, and glutathione-S-transferases, which render xenobiotics excretable through bile or urine. Third, stimulated AhR influences expression of organic anion/cation transporters, which affect transport of xenobiotics through the cell membrane. Overall, the alterations in expression levels of these genes are supposed to help detoxify harmful xenobiotics, and thus regarded as an adaptive mechanism. However, in our mouse experiment, plasma samples from the MMI-treated wild-type and AhR-deficient mice exhibited comparable AhR-stimulating activity (Fig. 7A), suggesting that activation of AhR by 3-methyl-2-thiohydantoin did not facilitate metabolism or disposal of 3-methyl-2-thiohydantoin itself, at least to an extent detectable in our reporter gene assay.
It has been reported that classical AhR agonists such as TCDD and PCBs cause hypothyroidism (19,20), by directly inhibiting thyroid hormone synthesis in thyrocytes (21) or by enhancing the thyroxine-conjugating UGT activity in the liver (22). Therefore, a question may be raised as to whether the serum AhR-stimulating activity observed in MMI-treated patients might be involved in the antithyroid action of MMI. Because plasma thyroxine concentration clearly declined after administration of MMI in AhR-deficient as well as wild-type mice (Fig. 7B), it can be safely concluded that MMI suppresses thyroid function even without activation of AhR. However, further experiments are needed to determine precisely to what extent the MMI-induced AhR stimulation contributes to the reduction of plasma thyroxine levels.
A possible concern for patients and physicians would be whether AhR activation is involved in any of the adverse effects of MMI, such as agranulocytosis, hepatobiliary damage, and skin rash. Many of these adverse effects can be observed in PTU-treated patients as well, indicating that they may not necessarily result from AhR stimulation. On the other hand, because TCDD or PCBs may cause neutropenia, hepatic injury, and skin symptoms (23 –28), one cannot eliminate the possibility that some of the adverse effects of MMI might be attributed, at least in part, to AhR activation. One adverse effect that is documented as possibly related to MMI, but not PTU, is the so-called MMI embryopathy comprising aplasia cutis, choanal or esophageal atresia, and characteristic facial features (29). Although similar malformations have not been reported in fetuses born from mothers exposed to TCDD or PCBs, AhR does play an important role in development and prenatal exposure to TCDD, or PCBs may affect fetal growth and immunological and neurological conditions (30). With the knowledge that AhR is activated by MMI administration, the possible effects of maternal MMI therapy on fetal development may be assessed more properly and strictly in the future.
Our finding may help elucidate some of the extrathyroidal actions of MMI that have not been commonly recognized so far. For instance, it has been reported that in a Graves' patient treated with irinotecan for colorectal cancer, plasma concentrations of the active irinotecan metabolite SN-38 were elevated by MMI administration independently of his thyroid hormone levels, and that this could be ascribed to induction of UGT1A1 by MMI (31). This phenomenon may be explained, at least in part, by our finding, because AhR agonists are known to upregulate UGT1A1 expression (32). Thus, whenever MMI is used for treatment of hyperthyroidism or for animal experiments, we need to consider effects of AhR activation on expression of different genes and metabolism of coadministered drugs. Based on our findings, more extrathyroidal actions of MMI as an AhR activator may be unraveled through further basic and epidemiological studies.
In conclusion, it has been found in humans and mice that treatment with MMI elicits serum AhR-stimulating activity, which is presumably ascribed to an MMI metabolite 3-methyl-2-thiohydantoin. Because we did not test serum samples from subjects exposed to classical AhR activators such as TCDD, PCBs, etc., it is difficult to tell how potent MTH-glycine is as an AhR activator, compared to TCDD or PCBs. Further study is required to assess the clinical impact of the dioxin-like action of 3-methyl-2-thiohydantoin and to determine the significance of the differences between MMI and PTU observed in this study.
Footnotes
Acknowledgments
We thank Ms. Kazue Murata, Ms. Mineko Fujita, and Ms. Yoshie Fujita for their assistance in laboratory work; Dr. Yoshiaki Fujii-Kuriyama and the RIKEN BRC for providing the AhR-null mice; and the animal facility staff for taking care of the mice.
Disclosure Statement
The authors declare that no competing financial interests exist.