
Abstract
Background:
Thyrotropin-secreting pituitary adenomas (TSHomas) are an extremely rare cause of hyperthyroidism. Up to now there are only few cases reported in the pediatric age range. Thefirst therapeutic option is surgical resection, whereas medical treatment with somatostatin analogs has been reported only in cases wherein surgery was unsuccessful.
Patient Findings:
A 13-year-old girl was referred to our clinic for incidental finding of increased circulating free thyroid hormones in the presence of detectable TSH concentrations. She had no signs/symptoms of thyrotoxicosis. Resistance to thyroid hormone was excluded due to the lack of TSH response after thyrotropin-releasing hormone (TRH) stimulation test. Cerebral magnetic resonance imaging showed the presence of a large pituitary macroadenoma, with intra- and suprasellar extension. We decided to treat this patient with somatostatin analog as a first-line therapy because of high surgery risks due to the tumor dimensions. The response to medical treatment was excellent, with rapid and significant tumor shrinkage. No major side effects were reported. The patient developed central hypothyroidism that was corrected with L-thyroxine therapy.
Summary:
We report the first pediatric case of TSHoma treated with somatostatin analog as a first-line therapy. The diagnosis was challenging because of the insidious and asymptomatic presentation of the tumor.
Conclusions:
We conclude that somatostatin analogs should be considered as first choice, bridge-to-surgery treatment in young patients, in order to reduce neurosurgical complications and prevent hypopituitarism during pubertal development.
Introduction
Failure to recognize the presence of a TSHoma may result in dramatic consequences, such as neurological and endocrine complications related to the tumor mass-effect or improper thyroid ablation that may cause the pituitary tumor volume to further expand.
The first therapeutic option is neurosurgery, but complete removal of the tumor is reported in 30–50% of the patients. Medical treatment with somatostatin analogs leads in most cases to hormone normalization, while tumor shrinkage is seen in about 50% of patients (2). In the pediatric age range, the use of somatostatin analogs has only been reported in two cases, both postpubertal 16-year-old boys in whom surgery was unsuccessful.
We report the case of a 13-year-old girl with asymptomatic TSH-secreting pituitary macroadenoma. She had no signs/symptoms of hyperthyroidism and she denied neurological disturbances, thus representing a diagnostic challenge. Because of the high surgery risks due to the tumor dimensions, she started somatostatin analog treatment with rapid significant tumor shrinkage. To our knowledge, this is the first report of first-line medical treatment with a somatostatin analog for a TSHoma in a pediatric patient.
Patient
A 13-year-old Caucasian girl had routine blood tests for transient episodes of palmar-plantar rash during fever. Her serum free thyroxine (fT4) (3.68 ng/dL, reference range [RR] 0.9–1.7) and free triiodothyronine (fT3) (13.6 pg/mL, RR 2.57–4.43) levels were elevated, in the presence of slightly elevated TSH (5.27 μIU/mL, RR 0.27–4.2).
The girl was referred to our clinic for further evaluation. She presented in good clinical condition, with regular growth (weight 55.4 kg, 50th–75th percentile; height 164.5 cm, 75th–90th percentile), blood pressure of 125/60 mmHg, and heart rate of 87 bpm. Her sexual development was at Tanner stage IV for pubic hair and Tanner stage III for breast; she had not yet reached menarche. Physical examination revealed that the thyroid gland was not enlarged. She denied symptoms of hyperthyroidism, except for occasional episodes of palpitation; she did not report neurological disturbances. There was no family history of thyroid disease.
Our laboratory confirmed hyperthyroidism (fT4 3.54 ng/dL, RR 0.7–1.7; fT3 9.25 pg/mL, RR 1.6–5.2) with unsuppressed TSH and negative thyroid autoantibodies. Thyroid ultrasonography showed normal dimensions and echogenicity of the gland. The thyrotropin-releasing hormone (TRH) stimulation test performed with intravenous Relefact® TRH 200 μg revealed an absent response of TSH (Table 1), not supporting the alternative diagnosis of resistance to thyroid hormone (RTH), while suggesting the presence of a TSH-secreting adenoma. Basal hormonal evaluation showed normal pituitary function.
Tested with Relefact® TRH 200 μg intravenous.
TRH, thyrotropin-releasing hormone; fT3, free triiodothyronine; fT4, free thyroxine.
The patient underwent cerebral magnetic resonance imaging (MRI) that showed the presence of a large pituitary macroadenoma measuring 2.8 cm × 2.5 cm × 2.9 cm, with intra- and suprasellar extension (Fig. 1). The tumor caused compression of the optic chiasm and the optic nerves in their prechiasmatic portion, with consequent moderate prechiasmatic left optic neuropathy (confirmed by visual field examination). The macroadenoma also had a portion of it lying over the sphenoidal planum with minimal compression on the posterior medial frontal convolutions. Due to the large dimensions and the shape of the tumor, complete surgical removal by transsphenoidal approach was considered to be unlikely.
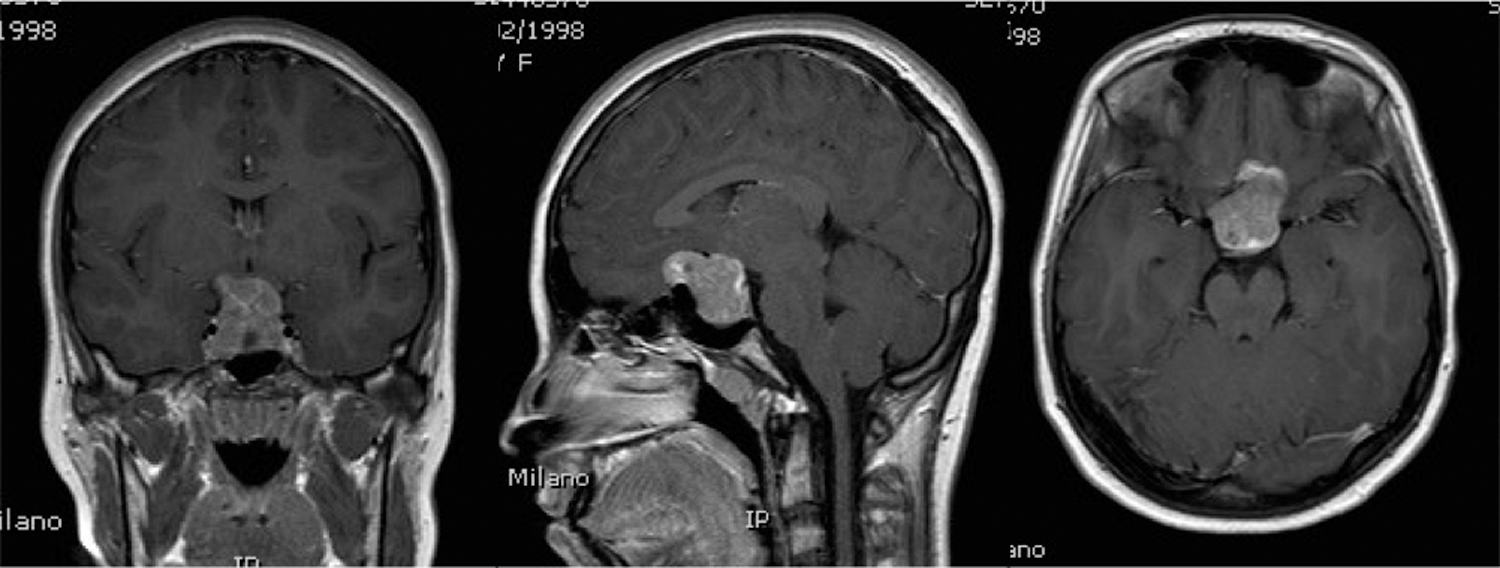
Magnetic resonance imaging at diagnosis, showing a large pituitary macroadenoma with intra- and suprasellar extension.
A single subcutaneous administration of octreotide 100 μg was associated with good suppression of TSH, consisting in a reduction of more than 50% (Table 2). Therefore, treatment with subcutaneous octreotide 100 mg three times a day was started. The patient did not show side effects, except for transitory mild abdominal discomfort. After few days she was discharged home with long-acting somatostatin analogs (intramuscular lanreotide 120 mg every 28 days).
Tested with octreotide 100 μg subcutaneously.
After 2 months of treatment she developed central hypothyroidism (TSH 0.486 μIU/mL, RR 0.25–5; fT4 0.63 pg/mL, RR 0.7–1.7; fT3 1.9 pg/mL, RR 1.6–5.2), and replacement therapy with L-thyroxine was started (50 μg/daily).
After 4 months of lanreotide treatment the MRI was repeated, showing a significant reduction of the TSH-secreting pituitary adenoma (1.5 cm×2 cm×2 cm) (Fig. 2). The optic neuropathy due to compression of the adenoma improved from moderate to mild. fT4 and fT3 were normal with L-thyroxine treatment (respectively, 1.1 ng/dL, RR 0.7–1.7 and 2.39 pg/mL, RR 1.6–5.2).

Magnetic resonance imaging
Because of the good response to medical therapy, absence of side effects, and age of the patient, we decided to continue lanreotide (120 mg once a month) and L-thyroxine (50 μg/daily) and to obtain an MRI every 6 months.
Discussion
Up to now about 10 cases of TSHoma in pediatric age have been reported in literature (3 –12) (Table 3). Like in adults, the majority of them are macroadenomas (9/10, 90%). Investigations leading to a diagnosis were undertaken for signs and symptoms of thyrotoxicosis or neurological features due to the pituitary mass. All patients underwent surgery with variable outcomes. Medical treatment with somatostatin analogs was used in only two patients, both 16-year-old boys, and as second-line treatment after unsuccessful surgical procedures.
TSS, transsphenoidal surgery; TCS, transcranial surgery; N/A, not available.
The first remarkable feature of our patient is that she was completely asymptomatic despite confirmed values of fT3 and fT4 more than double the normal upper limit. She denied symptoms of thyrotoxicosis (tachycardia, insomnia, attention deficit, hyperactivity, nervousness, etc.) and no alterations of growth and pubertal development could be detected. Thyroid function tests were performed for other reasons (as a routine screening after prolonged urticaria), with the incidental finding of increased circulating free thyroid hormones in the presence of detectable TSH concentrations. These biochemical features are characteristic of TSHomas, but they are also found in the RTH, in which there is a variable tissue hyporesponsiveness to thyroid hormone due to a defect in the thyroid hormone receptor beta gene (13). Usually, the main difference between these two syndromes consists of the presence of signs and symptoms of hyperthyroidism in patients with TSHoma. These are usually milder than in Graves' disease, while RTH patients are in general euthyroid and asymptomatic.
More than 90% of patients with TSHoma have a goiter, but this was also missing in our case (5). The following laboratory findings help to better distinguish TSHoma from RTH (2): (i) the serum alpha-subunit concentration is normal in RTH but often high in patients with TSH-secreting adenomas; (ii) the serum TSH concentration increases in response to TRH in patients with RTH, but not in most patients with TSH-secreting adenomas; (iii) patients with RTH are more likely to have a fall in serum TSH concentrations in response to T3.
The clinical presentation of our patient led us to first suspect RTH. In consideration, however, of the negative family history of thyroid disorders and the long time and costs needed for a genetic analysis of the TRβ gene, we decided to first perform a TRH test.
The feasibility of this test with regard to cost and test duration makes it a good tool to select the patients that should undergo imaging of the pituitary. Our case highlights the importance of also investigating the pituitary in the absence of clear neurological symptoms. Thus, our patient was found to have a macroadenoma of almost 3 cm diameter despite the fact that she had only slight visual field defects of which she was unaware.
Pituitary surgery is usually considered the first-line therapy for TSHomas. Trans-sphenoidal resection, however, results in cure in only about one-third of patients, mostly because the majority of the tumors are macroadenomas that often have hard consistency because of fibrosis (14). Also among the few pediatric cases, surgical remission was achieved in only 3 of 10 patients. A significant therapeutic advance was the introduction of somatostatin analogs, acting on the inhibitory somatostatin receptors expressed by almost all TSHomas. This medical treatment leads to free thyroid hormones normalization in 95% of cases and tumor size decrease in 52% (2). It is usually used after unsuccessful surgery, less frequently as first choice in patients with inoperable lesions. To our knowledge, there are only two reports of its use in pediatric cases of TSHoma: both patients were postpubertal boys who previously underwent unsuccessful surgery. The major adverse effects of somatostatin treatment are transient abdominal discomfort and gallstones. The most important risks for young patients, however, are possible decreases in pituitary growth hormone secretion and insulin secretion. Otherwise, pituitary surgery itself carries the potential risk of causing postoperative hypopituitarism.
The dimensions and especially the anterosuperior extension of our patient's tumor made the probability of complete surgical eradication by a transsphenoidal approach very low. The option of first-line treatment with long-acting somatostatin analogs was thoroughly discussed with the patient and her parents. The definite decision was taken in consideration of the favorable results of the octreotide suppression test. Long-term treatment was well accepted and tolerated by our patient, without significant side effects and with regular progression of pubertal development. Stable normalization of thyroid function was achieved after L-thyroxine treatment. The MRI performed after 4 months of treatment showed striking reduction of the tumor size, leading to potential better surgical outcomes with a transsphenoidal approach. Comparing the risks of surgery to the good response to medical treatment, and in agreement with the family, therapy with long-acting somatostatin is currently being continued.
In conclusion, while this disease is extremely rare, it highlights the importance of a prompt and correct diagnosis of TSHoma, even in pediatric age. It should be noted that clinical presentation might vary, with overlap with more benign disorders like RTH. In patients with increased circulating free thyroid hormones and detectable TSH concentrations, we recommend a TRH-test and then a cerebral MRI to exclude.
Somatostatin analogs represent not only an effective second-line therapy when cure is not reached by surgery. They should be considered also as first-choice treatment if the risks of surgery are elevated and the probability of cure is low. In particular, younger patients could benefit from medical treatment in order to avoid risks of hypopituitarism during pubertal development. It is crucial to first perform an octreotide suppression test as was done in our patient.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.