
Abstract
Background:
Postpartum thyroiditis (PPT) is characterized by the development of postpartum thyroid dysfunction, which may occur up to 12 months after delivery. The syndrome usually presents with transient thyrotoxicosis, followed by transient hypothyroidism. The association of this condition with resistance to thyroid hormones (RTH) has never been described.
Patient Findings:
In this report, we describe a 30-year-old patient affected by RTH due to a novel p.V283A thyroid hormone receptor-β (THRB) heterozygous mutation in exon 8, which affects the ligand-binding domain, never before described in literature. A simple polymorphism was excluded through screening of 100 healthy controls.
Summary:
The patient became pregnant twice (in 2008 and in 2009) and developed PPT after both deliveries. Two months after her first pregnancy and one month after her second pregnancy, she presented with severe endogenous thyrotoxicosis and concomitant suppressed thyrotropin (TSH) levels, which represents an unusual finding in patients affected by RTH. Other causes of hyperthyroidism were excluded. After the hyperthyroid phase, she became hypothyroid (TSH >75 mU/L and low free-thyroxine and free-tri-iodothyronine levels), and eventually returned to her usual euthyroid status. During the course of PPT, no specific treatment was required, except for β-blockers used to treat tachycardia during the hyperthyroid phase.
Conclusions:
We report a unique case of a woman affected by RTH, due to a novel mutation V283A in THRB, who experienced PPT with a severe thyrotoxic phase after both her pregnancies. The association between RTH and PPT has never been reported in the literature. In particular, the marked suppression of TSH occurring when levels of TH are particularly elevated is not a frequent condition during RTH.
Introduction
PPT can be associated with the presence of thyroid antibodies (Ab) and ultrasonography (US) features suggestive for autoimmune thyroiditis, strongly supporting an autoimmune origin of this condition. Indeed, it is believed that PPT is the result of an immunological postpartum rebound after the partial immunosuppression occurring in pregnancy. In fact, the presence of anti–thyroid peroxidase (TPO) or anti-thyroglobulin (Tg) Ab in the first trimester of pregnancy increases the chances of developing PTT by 33%–50%, compared to 0%–5% in the absence of thyroid Ab (2).
While cases of PPT have been described in association with comorbidities such as autoimmune hypophysitis (3,4), the association of PPT and thyroid hormone resistance (RTH) has never been reported so far.
Here we describe the case of a 30-year-old woman affected by RTH, due to a novel p.V283A thyroid hormone receptor-β (THRB) mutation, who developed PPT after both her pregnancies. In addition to the association between these two conditions, this case represents a unique model of the pattern of the thyroid function tests in RTH during both phases of transient-severe endogenous thyrotoxicosis, in which we could observe a marked suppression of thyrotropin (TSH), an infrequent condition in RTH.
Patient
A 30-year-old Caucasian woman was referred to us in September 2007 due to clinical symptoms of tachycardia, anxiety, generalized weakness, and nervousness. While her medical and pregnancy history was unremarkable, her family history revealed the presence of hyperthyroidism with inappropriate TSH secretion. On physical examination, the patient was normocardic (88 beats/min) and she had mild tremor of her extremities. No proptosis was present. Thyroid function tests were suggestive for hyperthyroidism with hyperthyrotropinemia: TSH, 5.9 mU/L (normal values [n.v.], 0.35–2.8); free thyroxine (FT4), 29.1 pg/mL (n.v., 8.5–15.5); free tri-iodothyronine (FT3), 4.8 pg/mL (n.v., 2.3–4.2). Tg-Ab and TPO-Ab were also elevated, while anti-TSH receptor (TR) Abs were negative. The thyroid US pattern was suggestive for thyroiditis with a calculated thyroid volume of about 35 mL and diffuse hypoechogenicity, without increased vascularity. TSH stimulation after exogenous TRH revealed an adequate response (TSH peak after TRH 17 mU/L), a finding suggestive for RTH. Pituitary magnetic resonance excluded the presence of an adenoma.
The patient underwent genetic testing for RTH, and a novel heterozygous mutation g.361470T>C (NC_000003) resulting in an amino acid substitution p.V283A was revealed in exon 8 of the THRB gene.
The woman did not receive any treatment for her condition, except low-dose atenolol (25 mg/day), and underwent periodic follow-ups.
In January 2008, the patient was re-evaluated in the first month of pregnancy. She stopped β-blocker therapy and started a careful monthly monitoring of her thyroid function that persistently showed high FT3 and FT4 levels and concomitant nonsuppressed TSH levels (Table 1). These biochemical features were in agreement with condition of RTH, and did not require any medical treatment until delivery. Two months after delivery (full-term male neonate of 3150 g, with normal thyroid function at birth: TSH, 13.4 mU/L; FT3, 5.6 pg/mL; FT4, 12.0 pg/mL) in November 2008, the patient, who elected not to breastfeed, developed severe thyrotoxicosis (TSH, <0.03 mU/L; FT3, 15.2 pg/mL; FT4, 72 pg/mL), again requiring β-blocker therapy. During this period, the patient showed classic symptoms of hyperthyroidism, in particular general fatigue, palpitations, and heat intolerance. Graves' disease was excluded based on negative TR-Ab and low radioactive iodine uptake (<1% at 24 hours). The physical examination was negative for pain in the anterior region of the neck, and blood count and inflammatory parameters were normal (erythrocyte sedimentation rate, 9 mm/h; C reactive protein, <0.5 mg/L). Thyroid US confirmed thyroiditis without increased vascularity. Taken together, all data were suggestive of PPT. Two months later, consistent with the typical course of PPT, the patient went through a hypothyroid phase (TSH, 102 mU/L; FT4, 6.5 pg/mL; FT3, 3 pg/mL), which showed a progressive spontaneous remission without any levothyroxine (LT4) therapy. Clinical symptoms also improved. Eight weeks after the diagnosis of hypothyroidism, the patient returned to her usual euthyroid state (FT3, 4.3 pg/mL; FT4, 16 pg/mL) with the persistence of inappropriately elevated TSH values (9 mU/L) consistent with the pre-pregnancy profile (Fig. 1A). Further periodic evaluations showed that, despite high Tg-Ab and TPO-Ab levels, there was no evolution to permanent hypothyroidism.
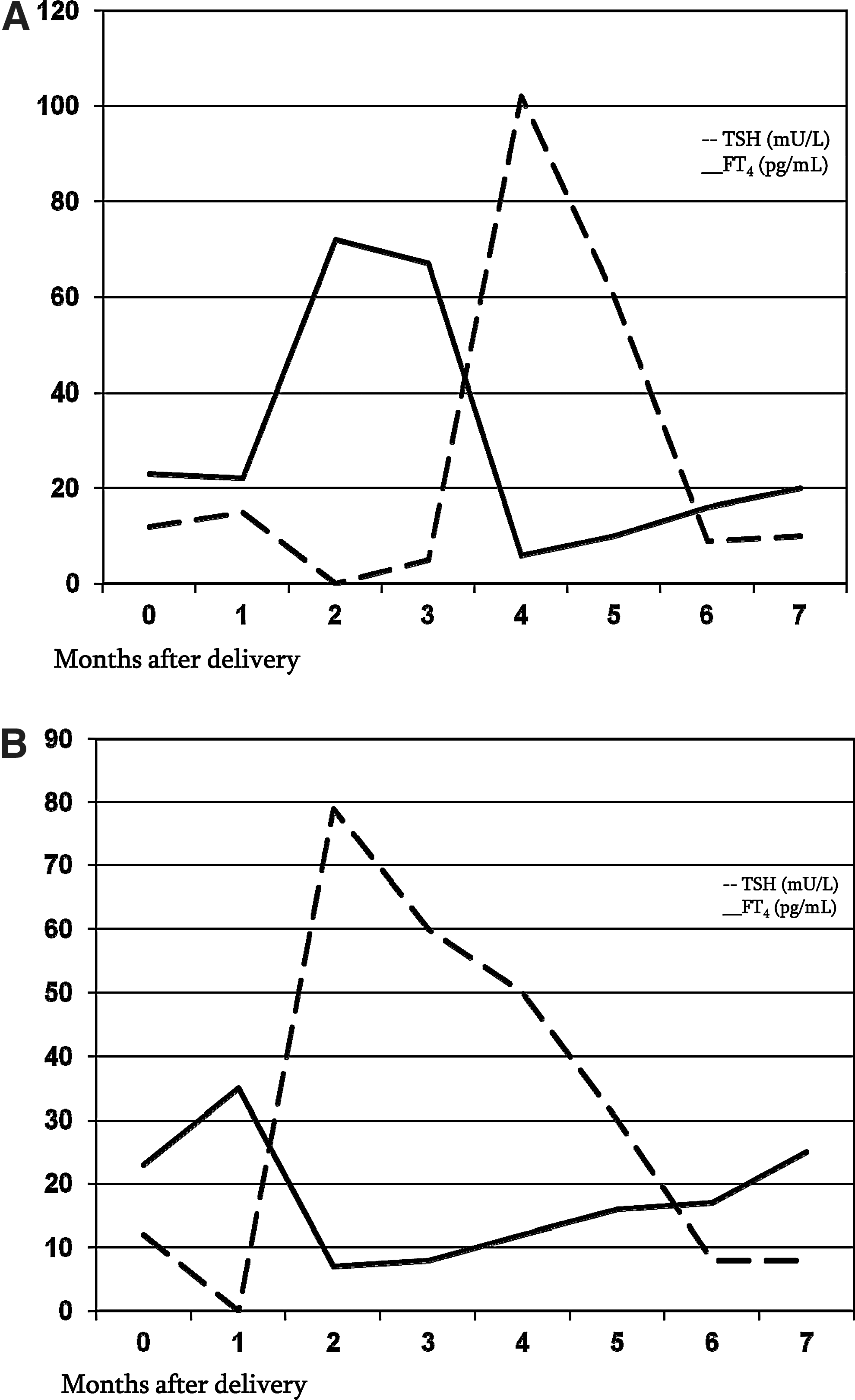
The thyroid function pattern of a patient affected by resistance to thyroid hormone who developed a postpartum thyroiditis after both of her pregnancies. In her first pregnancy
FT3, free triiodothyronine; FT4, free thyroxine; TSH, thyrotropin; n.v., normal values.
In November 2009, the patient was pregnant again. As in the first pregnancy, she started a careful monthly evaluation of her thyroid function which showed only slightly elevated FT4, FT3, and TSH levels, for which she did not require any treatment (Table 2). One month after delivery (full-term male neonate of 2900 g with normal thyroid function at birth: TSH, 11.4 mU/L; FT3, 5.3 pg/mL; FT4, 14.0 pg/mL), in September 2010, she again elected to not breastfeed and developed moderate to severe thyrotoxicosis (FT3, 10 pg/mL; FT4, 35 pg/mL) with suppression of her TSH levels (TSH <0.03 mU/L). During this period, thyrotoxic symptoms were documented. To exclude Graves' disease and confirm PPT, thyroid radioactive iodine uptake, US, and TR-Ab evaluation were performed: the reduced radioactive iodine uptake (<1% at 24 hours), the normal vascularity on US, and negative TR-Ab were clearly suggestive of PPT. The patient was treated only with β-blockers, and her thyroid function was periodically monitored. The biochemical course of PPT that occurred after the second pregnancy was identical to that of the first pregnancy: 2 months after delivery, the patient became hypothyroid (FT3, 1.8 pg/mL; FT4, 6.7 pg/mL; TSH, 79 mU/mL), and her thyroid function and clinical status returned to her normal profile within 6 months after delivery. The latest evaluation, performed in February 2011, showed a complete recovery and return to the pre-pregnancy hormonal profile (FT3, 4.7 pg/mL; FT4, 17 pg/mL; TSH, 8.1 mU/mL) with persistence of positive anti-Tg-Ab and anti–TPO-Ab (Fig. 1B).
Gene analysis
After obtaining informed consent, the patient underwent genetic testing for RTH. Sequencing of the complete THRB gene, performed as previously reported (5), revealed a novel heterozygous mutation g.361470T>C (NC-000003) resulting in an amino acid substitution p.V283A in exon 8 of the gene. This mutation is localized in the region that encodes, together with exon 9 and exon 10, the ligand-binding domain of the receptor. This mutation has never been reported in the literature. For this reason, to exclude that p.V283A is a TRHB polymorphism, we screened 100 healthy controls (200 alleles) by high-resolution melting analysis (primer and conditions are available upon request). All screened subjects come from central and southern Italy. This screening was negative for the above-mentioned mutation, indicating a frequency of <1%. Unfortunately, the genetic testing on the siblings suspected to have the subject's same condition could not be performed due to their refusal to provide informed consent.
Discussion
RTH is a syndrome of reduced end-organ responsiveness to the thyroid hormone (TH), first described by Refetoff in 1967 (6). The biochemical features are characterized by elevated serum levels of FT4 and FT3 and inappropriately elevated TSH, while clinical manifestations can be extremely diverse because of variable degrees of hormone resistance, and differences in end organ response and compensatory mechanisms (7). In our patient, at the diagnosis, FT3, FT4, and TSH were all only slightly elevated, resulting in mild symptoms of hyperthyroidism. While it is difficult to differentiate the selective pituitary resistance from the generalized resistance syndrome, it is very likely that our patient was affected by a mild pituitary form of RTH. In this situation, a new balance was established and it was characterized by high FT4 and FT3 with a nonsuppressed TSH level. The patient developed more severe thyrotoxicosis during PPT, as shown by laboratory tests. RTH is caused in ∼90% of cases by a dominant mutation in the THRB gene. In patients affected by RTH, most of the identified mutations of the THRB gene are located in exon 9 and exon 10 (8) and only few mutations in exon 8 (9 –11). The link between RTH and THRB mutations was first reported in 1988, and >100 mutations have been described since then (12). In >85% of families with RTH, THRB mutations have been identified, of which 23% are considered de novo mutations. Interestingly, in ∼15% of families with RTH, no THRB mutations are detectable (non-TR RTH) (13,14). In our patient, the THRB molecular analysis showed a novel mutation in exon 8 (p.V283A)—which, with exon 9 and exon 10, encodes the ligand-binding domain and is likely the cause of the RTH. Further in vitro analyses are required to assess the functional consequences of this substitution.
Few descriptions of the outcome of pregnancies in women with RTH are available in the literature (15,16). Transient thyrotoxicosis associated with early gestation in patients with RTH has been reported (17). Some authors recommend the prenatal analysis of the THRB gene in affected families because of potential risks to the fetus associated with RTH (16). In fact, an unaffected fetus carried by an RTH mother may be suffering of a placental transfer of excess maternal TH. Despite that, based on the mild maternal elevation of FT3 and FT4, we did not perform prenatal THRB molecular analysis in the two fetuses, but we carefully monitored maternal thyroid function and fetal morphology through US during both pregnancies. No complications occurred during the pregnancies or in the postpartum periods.
In the absence of prospective data on a large number of RTH pregnancies, there are no clear guidelines on the management of such patients. Given the low frequency of this condition, it would be important that investigators share their own experience with the scientific community.
We also report the association between RTH and PPT observed in this case. PPT is an autoimmune disease: the exacerbation of an underlying autoimmune thyroiditis, worsened by the immunological rebound that follows the partial pregnancy-induced immunosuppression, is involved in the pathogenesis of this disorder.
The underlying mechanisms of PPT are not known. In subjects affected by this condition, a higher CD4/CD8 ratio has been described in the peripheral blood (2) and in the thyroid (18), as well as an elevation in activated T cells (19), suggesting an increase in the immune activity during and after pregnancy, which potentially could trigger PPT.
Another possible hypothesis is microchimerism, due to fetal immune cells crossing the placenta and homing to the maternal thyroid gland triggering an autoimmune reaction to the graft-versus-host reaction (20).
The presence of PPT in a patient with RTH has never been described so far. The most interesting feature in our patient is the recurrence of PPT, characterized in both occasions by marked suppression of TSH values during the phase of endogenous thyrotoxicosis.
In fact, in our patient with RTH, we observed the classic sequential phases of PPT: thyrotoxicosis, hypothyroidism, and recovery, the latter phase being characterized by the return to the status quo ante of mild hyperthyroidism with inappropriate TSH secretion.
The most important and peculiar observation is that during the severe endogenous thyrotoxicosis induced by the PPT, the patient showed an unexpected suppression of TSH (<0.03 mU/L) in spite of her underlying condition characterized by inappropriately elevated TSH levels. This laboratory picture has already been described in some patients with RTH affected by Graves' disease, in which autoimmune hyperthyroidism was able to cause a TSH suppression despite the underlying condition (TSH always ≤0.2 mU/L, with the lowest reported FT3 values of 7 pg/mL and FT4 of 24.5 pg/mL) (21 –24). Indeed, suppressed TSH levels in patients affected by RTH are not commonly observed nor achieved: it is well known that supraphysiological doses of LT4 therapy cannot achieve TSH suppression (with daily LT4 doses of ∼3 and ∼2 μg/kg, the TSH levels were 28 and 48 mU/L, respectively) (5). Suppressed TSH can be obtained only by administering supraphysiological doses of T3 (>50 μg daily) (25,26). It has also been described that treatment with supraphysiolocal doses of L-triiodothyronine, given as single doses daily, reduces the goiter size (27). The intracellular T3 concentration, both at the hypothalamic and pituitary level, is the major determinant of TSH secretion, including in patients affected by RTH. In PPT, the destruction of the thyroid follicles leads to a transient release of T3 and T4 from the damaged gland: the resulting endogenous thyrotoxicosis, and particularly the T3 toxicosis, appears to be able to override the mechanism of resistance at the hypothalamic–pituitary level, allowing the transitory suppression of TSH.
Footnotes
Disclosure Statement
All authors declare that they have no competing financial interests and no conflict of interest.