
Abstract
Background:
The nuclear factor kappa-B (NF-κB) proteins, a family of transcription factors found virtually in all cells, are known to play crucial roles in the growth of a number of human malignancies. The ability of NF-κB to target a large number of genes that regulate cell proliferation, differentiation, survival, and apoptosis, provides clues toward its deregulation during the process of tumorigenesis, metastatic progression, and therapeutic resistance of tumors.
Summary:
In addition to the signaling pathways known to be involved in thyroid tumorigenesis, such as the mitogen-activated protein kinase and janus kinase cascades, studies implicate the NF-κB pathway in the development of both less aggressive thyroid cancers, papillary and follicular adenocarcinomas, and progression to aggressive thyroid cancers, such as anaplastic adenocarcinomas. A constitutively activated NF-κB pathway also closely links Hashimoto's thyroiditis with increased incidence of thyroid cancers. The NF-κB pathway is becoming one of the major targets for drug development, and a number of compounds have been developed to inhibit this pathway at different levels in cancer cells. Some of these targets have shown promising outcomes in both in vitro and in vivo investigations and a handful of them have shown efficacy in the clinical setting.
Conclusions:
This review discusses the recent findings that demonstrate that the inhibition of NF-κB, alone or with other signaling pathway inhibitors may be of significant therapeutic benefits against aggressive thyroid cancers.
Introduction
NF-κB and its activation pathway
The NF-κB protein was first discovered as a DNA-binding protein that interacts with an 11-base pair sequence in the immunoglobulin kappa light-chain enhancer in B cells. Since its first discovery in 1986, the NF-κB cascade has been well recognized as a transcription factor, which can regulate the expression of a large number of genes that are critical for apoptosis, tumorigenesis, inflammation, and various autoimmune diseases (7). In mammals, the NF-κB family of transcription factors is composed of five distinct members: RelA (p65), RelB and c-Rel, NF-κB1 (p50 and its precursor protein p105), and NF-κB2 (p52 and its precursor protein p100). All these NF-κB members share a highly conserved 300 amino acid dimerization domain known as the Rel Homology Domain, which is required for binding DNA and enhances a variety of target genes. RelA (p65), RelB, and c-Rel contain another common carboxy-terminal structure called transactivation domain, which exerts a positive role in transcription complex formation by binding to numerous other proteins, such as transcriptional coregulators. Several different structural combinations of NF-κB dimers exist in the cytoplasm, in which p65/p50 is the most common heterodimer. These subunits can also combine as homodimers (p50/p50, p52/p52) and regulate target genes positively or negatively by binding the inhibitor of NF-κB (IκB) family member Bcl-3(8).
The NF-κB signaling pathway, which can be activated by both extracellular and intracellular stimuli, includes, the cell surface receptors (tumor necrosis factor receptor [TNFR], interleukin-1 receptor [IL-1R], Toll-like receptor 4 [TLR4], nucleotide oligomerization domain–like receptor [NLR], lymphotoxin beta receptor [LTβR], B-cell activating factor receptor [BAFF-R], receptor activator of NF-κB [RANK], etc.), the receptor signal adaptor protein (tumor necrosis factor [TNF], TNF receptor–associated factors [TRAFs], receptor-interacting proteins [RIPs]), IκB kinase (IKK) complex, IκB proteins, and dimers of NF-κB subunits. An IKK complex consists of three subunits, including IKKα, IKKβ, and IKKγ (also named NEMO). IKKβ can induce phosphorylation of IκBα at serine 32 and 36 and causes the subsequent degradation of IκBα. NEMO, as a regulatory subunit of the IKK complex, plays an integral role in the activation of NF-κB by modulating degradation of IκBα. Following a wide variety of external stimuli, the IKK complex is activated, which results in phosphorylation and ubiquitination of the IκB protein. After IκB protein degradation by the proteasome, bound NF-κB dimers are released and translocate to the nucleus. After further post-transcriptional modification in the nucleus, NF-κB is able to transcribe a selective subset of target genes controlling different aspects of cell physiology.
Thus, the activation of the NF-κB pathway can be triggered by various stimuli in a canonical (classical) and noncanonical (alternative) manner. The canonical pathway introduced by most physiological stimuli involves cell membrane receptors, such as TNFR, IL-1R, and TLR4, which are able to recruit the adaptors TRADD, TRAF2, and RIP in the cytoplasm and sequentially phosphorylate the IKK complex. Activation of the IKK complex leads to phosphorylation and ubiquitination of IκBα and releases the heterodimer p65/p50 from the cytoplasm leading to its nuclear translocation, which sequentially activates target genes. The activation of the noncanonical pathway triggered by CD40, RANK, LTβR, and BAFF depends on phosphorylation of IKKα, which is not necessary in the canonical pathway. Activation of IKKα sequentially processes p100 into p52, which dimerizes with RelBand and forms the NF-κB subunit to trigger transcription of target genes in the nucleus (9). The complete NF-κB pathway activation pathway is illustrated in Figure 1.
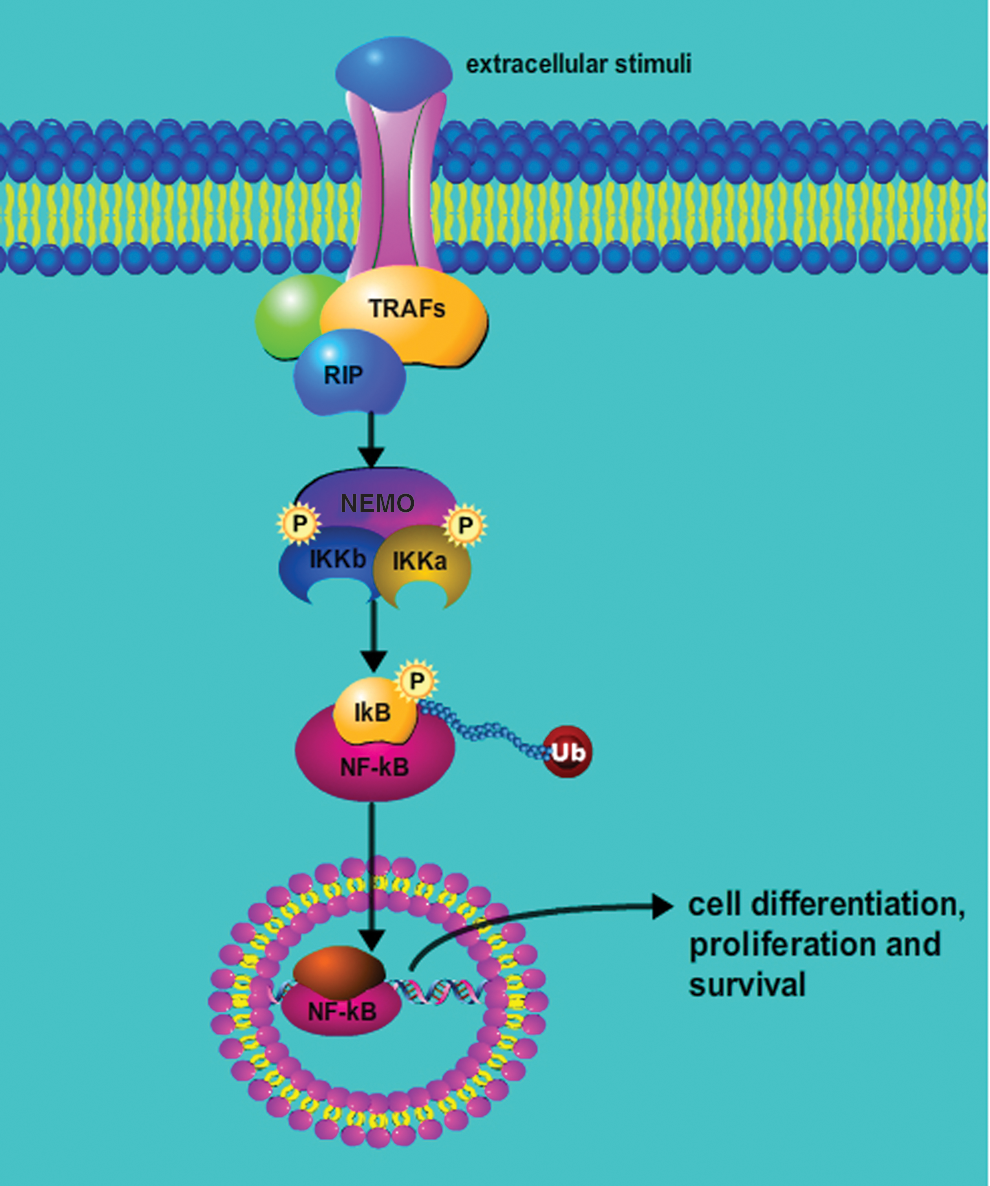
The NF-κB signaling cascade. In cells, NF-κB activation is triggered by extracellular stimuli, which transduce the signal via membrane receptors to sequentially phosphorylate the IKK complex. The IKK then phosphorylates IκBα, which is released from the NF-κB complex, ubiquitinated and degradated by the proteasomes. The released NF-κB heterodimers are able to translocate from the cytoplasm to the nucleus, where they bind to cognate DNA sequences and activate numerous target genes. In cancer cells, these NF-κB–induced genes are crucial in regulating cell proliferation, metastasis, survival, and therapeutic resistance. TRAF, tumor necrosis factor receptor–associated factor; RIP, receptor-interacting protein; NEMO, NF-κB essential modulator; P, proteasome; IKK, IκB kinase; IκB, inhibitor of NF-κB; NF-κB, nuclear factor kappa-B; Ub, ubiquitination. Color images available online at
Signaling pathways cross-talk with NF-κB and regulate its function
Multiple parallel signal transduction pathways are also involved in the activation of NF-κB. As a major regulator of cell survival, NF-κB can act synergistically with other important oncogenic signaling cascades to ultimately regulate tumorigenesis. The cross-talk between NF-κBand and these parallel pathways are well documented in a recent review article (10). In this article, we have emphasized the role of several of these important signaling pathways, including the c-Jun-NH2-terminal kinase (JNK) pathway, PI3K (phosphatidylinositol 3-kinase) pathway, and P53 pathway, which are known to play an essential role in the pathogenesis of thyroid cancer.
The JNK pathway (a signaling pathway that is known to promote apoptosis) and NF-κB have a common regulatory node via the TRAFs. External stimulus, such as TNFα, can target TRAF2, which can then activate both JNK and NF-κB signal pathways. The balance between JNK and NF-κB activities is considered crucial to determine the cell fate in response to external stimuli (11,12). Similarly, with IL-1R or TLR4 signaling, TRAF6 can induce TAK1 to trigger the activation of both of these two signaling pathways (13,14). Alternatively, NF-κB-induced gene products may also affect JNK activity. For instance, the ferritin heavy chain or manganese-superoxide dismutase blunts sustained JNK activity. Proteins inhibiting JNK signal activation, such as GADD45β, A20, and XIAP, can also be produced due to the activation of the NF-κB signal pathway (11). These signaling pathways and their cross-talk with NF-κB are depicted in Figure 2.
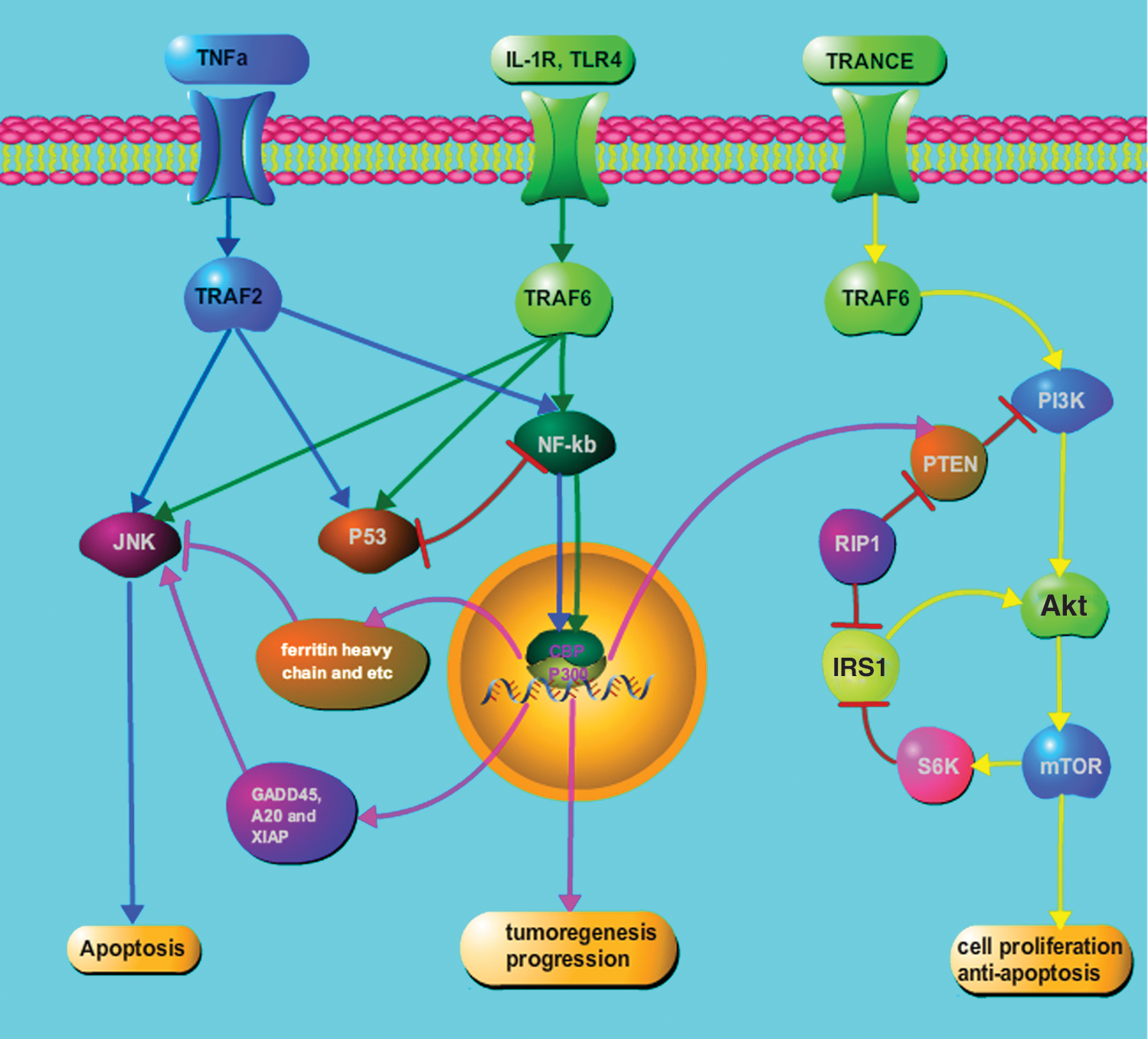
Cross-talk between NF-κB and parallel signaling pathways. Several second messenger signaling pathways can cross-talk with the NF-κB activation cascade. These multiple nodes of regulation of the NF-κB pathway are also suggested to be involved in the growth of aggressive thyroid cancers. Green lines show the NF-κB pathway, blue lines show the JNK pathway, and the yellow lines shows the PI3K pathway. The pink lines demonstrate how NF-κB activation can alter the functioning of these parallel signaling pathways in tumor cells. IL-1R, interleukin-1 receptor; TLR4, Toll-like receptor 4; TRANCE, TNF-related activation-induced cytokine; JNK, c-Jun-NH2-terminal kinase; GADD, growth arrest and DNA damage; XIAP, X-linked inhibitor of apoptosis protein; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homolog; mTOR, mammalian target of rapamycin. Color images available online at
The tumor suppressor PTEN, which acts as a negative regulator of the PI3K/Akt pathway, is downregulated by NF-κB to prevent apoptosis (15). In addition to the downregulation of PTEN by NF-κB, RIP1 activates PI3K/Akt through a mechanism involving NF-κB-mediated inhibition of the mTOR-S6K–IRS1 negative feedback loop (16,17). TNF-related activation-induced cytokine, a TNF family member, activates the PI3K/Akt pathway through a signaling complex involving TRAF6 (18), which is also involved in the canonical activation of NF-κB pathway. It was revealed (19,20) that the PI3K/Akt pathway can switch second messenger signaling from the occupied IL-1 receptor to NF-κB, and specific PI3K inhibitors can strongly inhibit both PI3K activation and NF-κB-dependent gene expression. Interestingly, both overexpression of the p110 catalytic subunit of PI3K and the PI3K-activated protein kinase Akt can induce p65/RelA-mediated transactivation.
The tumor suppressor P53 is known to be an important regulator of cellular apoptosis. Under certain stimuli, such as TNFα, various tumor cell lines elicit different responses, which depend predominantly on activation of the P53 and NF-κB pathways. Indeed, P53 is known to be a competitive inhibitor of NF-κBp65 that can function through the sequestration of the CREB-binding protein (CBP/P300), a cofactor required for activation of both the P53 and the NF-κB pathways (21). Huang et al. (22) showed that phosphorylation of CBP at serine 1382 and serine 1386 increases IKKα-induced HAT activity of CBP, which then enhances its binding capability with NF-κBp65 and reduces its binding to P53.
The aforementioned studies argued that NF-κB activation acts in concert and as part of a complex network of other signal transduction pathways, under both physiologic conditions and especially under diseased states, such as cancer. Therefore, systemic blockade of NF-κB may intervene with these other pathways and cause unexpected adverse effects. Thus, understanding the cross-talk with other pathways will be helpful to elucidate how to selectively inhibit NF-κB in cancer cells without influencing its normal physiologic functions in normal tissues.
The crucial role of NF-κB pathway in thyroid cancers
Over the past two decades, the diverse roles of NF-κB have been well documented in multiple carcinomas, including leukemia, lymphoma, head and neck squamous carcinoma, ovarian, prostate, colon, and breast cancers (23). These diverse effects have been linked to the activation of cell proliferation, anti-apoptosis, tumor progression, and invasion, as well as resistance to chemotherapy and radiotherapy (24,25). In most of the above cancers, although NF-κB has been primarily considered as a tumor-promoting factor, studies suggest that the induction of NF-κB signaling may actually have disparate effects in different thyroid cancers.
An earlier study had reported constitutive activation of NF-κB in a panel of thyroid carcinoma cell lines (4). With the exception of two lines (NPA and ARO) that were found not to be of thyroid origin, thyroid cancer cell lines included in this study were found to have significantly increased p65 mRNA and protein expression compared to normal thyroid cells, further complicating the role of NF-κB in thyroid cancers (26). Interestingly, by immunohistochemical staining using an anti-p65 antibody, tissue specimens from papillary, follicular, and anaplastic thyroid cancers were shown to have constitutively activated NF-κB (5,27), clearly implicating the importance of this family of transcription factors in different stages of thyroid cancers. A number of recent molecular studies also suggest the reason for this activated NF-kB levels.
Thyroid carcinoma comprises a group of different types of tumor cells with distinctive clinical and pathological characteristics that occur due to different genetic mutations involving specific oncogenes. The RET/PTC rearrangements, as well as BRAF and RAS mutations, are often seen in aggressive cancers. These genetic alterations potently activate the MAPK pathway, which can in turn cause NF-κB activation and oncogene-mediated progression and aggressive behavior of papillary thyroid carcinomas (28 –31). A common mutation found in the BRAF gene (i.e., BRAFV600E ) can also activate NF-κB, which occurs via the direct activation of BRAF, but is independent of ERK signaling (32). Neely et al. (33) recently reported that the RET/PTC-3 gene can also activate the canonical NF-κB pathway by stabilizing the NF-κB-inducing kinase (NIK) in both papillary thyroid carcinomas and in thyroid epithelial cells found in Hashimoto's thyroiditis. Loss of function of tumor suppressors, including PTEN downregulation, P53 mutations, and β-CATENIN, also contributes to the progression of thyroid cancer (34 –37). Inactivation of PTEN in thyroid cancer can oppose the activation of PI3K/Akt pathway, thus, in turn, increasing NF-κB activity and accelerating tumor progression (15,38). PI3K/Akt can also downregulate the FOXO3a activity, which is a central mediator of cell cycle and apoptosis (39). In contrast with other carcinomas, mutations in P53 and its family members P63 and P73 are not common in early stages of thyroid cancer, but are mainly manifested in the poorly differentiated and aggressive phenotype of thyroid cancers (40 –42). Indeed, the downregulation of cyclin B1 by NF-κB inhibition in the 8505C-ATC cell line showed P53-induced cell cycle arrest through an increase in the level of p21WAF1 (43). Kroll et al. (44) were the first to report that the PAX8-PPARγ fusion, which results from a chromosomal translocation of t (2; 3) (q13; p25) in human thyroid follicular tumors, lacks the ability of wild-type PPARγ to inhibit NF-κB activation and results in the activation of cyclin D1, which causes repression of critical genes involved in apoptosis (45).
The relationship between inflammation and cancer was first noted by Rudolf Virchow in 1863 (46). Subsequent studies provided the connection between gastritis associated with chronic Helicobacter pylori infection and the increased occurrence of gastric cancers. Further studies have also linked colonitis with colon cancer, cholangitis with cholangiocellular carcinoma, and hepatitis with hepatocellular carcinoma (47). Extensive studies have proposed further molecular mechanisms, which show that the NF-κB pathway is a major link between inflammation and neoplastic transformation, as well as cancer progression. The inflammation process chronically activates NF-κB. This stimulates the expression of cytokines, chemokines, growth factors, and protease cascades, which favors initiation and progression of tumors, and provides a niche for malignant transformation. Arif et al. (48) observed that Hashimoto's thyroiditis, a chronic inflammatory condition, shares many common morphological features and immunohistochemical staining patterns as observed in thyroid cancers. Asioli et al. (49) also found that Hashimoto's thyroiditis shows atypical nuclear features, including prominent nuclear grooves, enlarged overlapping nuclei, and nuclear clearing, which are often evident in thyroid cancers. The coexistence of Hashimoto's thyroiditis in PTC cases has been associated in some studies with unfavorable clinical outcomes compared with those without Hashimoto's thyroiditis (48 –52), further strengthening the link between chronic inflammation and progression to thyroid carcinomas.
The oncogenes found to be relevant in different signaling pathways in thyroid cancers, such as RAS, RET, and BRAF, can recruit and activate inflammatory cells and reduce anticancer immune surveillance. With the loss of PTEN function in thyroid cancers, downstream PI3K/Akt pathway activation is observed in both Hashimoto's thyroiditis and thyroid cancer (34,51). As previously discussed, the NF-κB pathway can be activated by PI3K/Akt, which can then promote angiogenic and metastatic gene expression (53). The RET/PTC3 (RP3) fusion gene product is an oncogenic protein that is frequently expressed in papillary thyroid carcinoma accompanied by Hashimoto's thyroiditis. The RP3 genetic background is also known to induce the secretion of proinflammatory mediators, including granulocyte-macrophage colony–stimulating factor and macrophage chemotactic protein 1, and suppresses innate immunity against cancers, which results in the onset and progression of thyroid cancer (54,55). Indeed, it has been shown that the RP3 oncogene can activate the canonical NF-κB pathway by stabilizing NIK (33).
NF-κB inhibitory strategies as targets for thyroid cancer therapy
Due to the important role played by NF-κB, inhibitors of this pathway may be promising targets against thyroid cancers (Fig. 3). The sustained activation of NF-κB has also been associated with tumor recurrence following both chemotherapy and radiotherapy. Therefore, adjunct treatment with NF-κB inhibitors may be of great benefit to reverse therapeutic resistance (56 –58). Numerous NF-κB inhibitors are currently being tested in different laboratories and several of them are in clinical trials. These compounds can inhibit the NF-κB activation pathway at different levels, such as at the activation of IKK complex, stabilization of the IκBα, activation of NF-κB inhibitory proteins, activation of the proteasome cascade, and lastly, the targeting of the transcriptionally active NF-κB heterodimers, such as the RelA subunit. In addition, NF-κB inhibitory strategies may be feasible by targeting signal pathways from upstream effectors, such as TRAFs and NIK, as well as the inhibition of downstream factors, for example, direct blockade of NF-κB binding to DNA. Furthermore, parallel inhibition of NF-κB-independent pathways and its cross-talk with these pathways may provide more efficient and long-lasting therapeutic outcomes for tumors, especially due to the multiple mechanisms via which NF-κB and its regulatory signaling events might ultimately affect cancer initiation, progression, and recurrence/resistance (32,43).
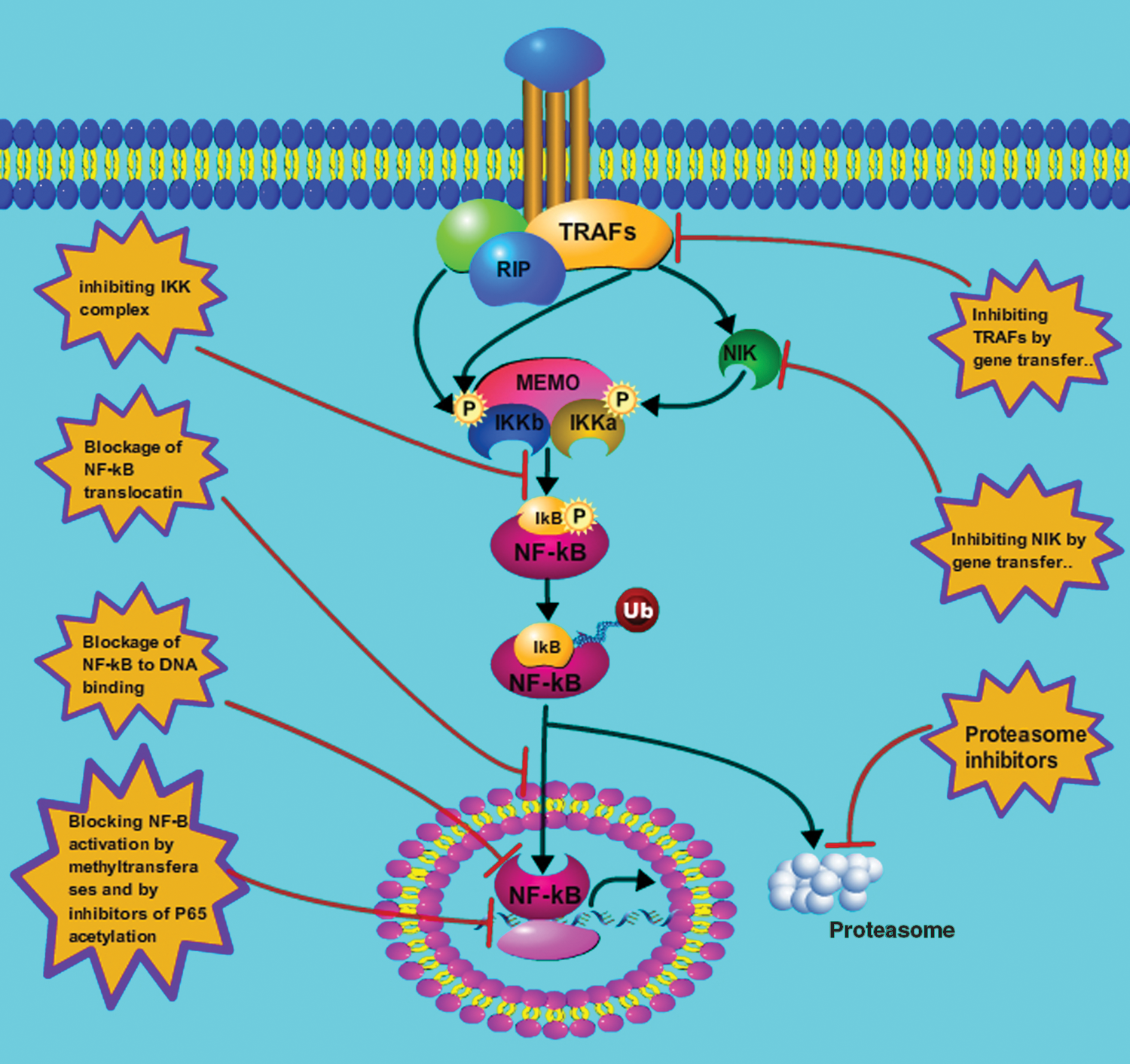
NF-κB inhibitors as targets for thyroid cancer therapy. The content in the stars show numerous strategies being used to inhibit the activation of NF-κB at different levels, such as the IKK complex, the IκBα, inhibitory proteins, the proteasome, and the RelA subunit of the transcriptionally active heterodimers. Color images available online at
Notably, it is well established that TRAFs transduce signals that emanate from most members of the TNFR superfamily and the IL-1 receptor/Toll-like receptor super family; these are able to activate both canonical and noncanonical NF-κB pathway (59,60). A recent study has shown that dominant-negative TRAF2 mutants or downregulation of TRAF2 achieved by siRNA can significantly inhibit NF-κB activation in response to TNF-α (61). Similarly, in human 293 cells, IL-1-induced NF-κB activation could be blocked by using a dominant-negative mutant of TRAF6 (62). It should also be noted that, in addition to suppressing the NF-κB pathway, inhibition of TRAFs resulted in the inactivation of the JNK pathway, as well. The NIK protein that is induced by several NF-κB activators, such as TRAF-2, TRAF-6, CIAP1, and CIAP2, was found to activate NF-κB through the noncanonical pathway. This activation of NF-κB was inhibited in NIK-deficient murine embryonic fibroblasts, despite an increase in NIK induced by the RET/PTC (RP3) gene product (33). Also, a kinase-dead mutant of IKKα, an RIP mutant lacking a kinase domain, or direct suppression of IKKα by siRNA technology, were able to significantly block the PDCasp8/10-mediated NF-κB activation (63). Since the activation of NF-κB requires the activity of the IKK complex (IKKα, IKKβ, and IKKγ), repression of IKKs has been recognized as another strategy to inhibit NF-κB activation by increasing the accumulation of IκBα. Many pharmacologically available anti-inflammatory drugs, such as NSAIDS, manifest their inhibitory effects on NF-κB activation via this pathway. These include aspirin, ibuprofen, mitricoxide, prostaglandin, sanguinarine, 4-hydroxy-2-nonenal, curcumin, and ursolic acid, which function by suppressing the phosphorylation of IκBα via the IKK complex (64 –71). Of note, several highly selective inhibitors of IKKβ, such as PS1145 and BAY 11-7082, which suppresses proinflammatory stimuli-induced IκBα phosphorylation, are under investigation for the treatment of different carcinomas (72). Furthermore, since the proteasome is necessary to degrade the IκB inhibitory subunit after its phosphorylation and ubiquitination in the cytoplasm (73), proteasome inhibitors are potent drugs to block NF-κB activation by preventing 26S proteasome from degrading IκBα. Therefore, several proteasome inhibitors, such as MG132, lactacystin, epoxomicin, MLN519, and bortezomib, are being evaluated for their therapeutic effects against diverse human cancers. Indeed, bortezomib has been clinically approved for the therapy of several different types of solid tumors, and its use in combination therapies has been proven to enhance sensitivity to both chemotherapy and radiotherapy (72,74 –79).
The most direct strategy for blocking NF-κB activation is to block its binding to its cognate nucleotide sequences in the enhancer regions of numerous genes. Oligonucleotide decoys (OD), short synthetic fragments of DNA or RNA, can competitively inhibit NF-κB binding to these complementary regions of specific DNA, and have been very successful in this respect. The first reported OD was a double-stranded oligonucleotide, which was able to compete with the endogenous DNA for NF-κB binding (80). Direct injection of the NF-κB OD into colon carcinomas implanted in mice was shown to inhibit cachexia (81). Some sesquiterpene lactones have also been reported to inhibit NF-κB binding by interacting with Cys-38 in the DNA-binding loop of RelA and through an analogous Cys residue in the DNA-binding loops of p50 and c-Rel (82,83). These sesquiterpene lactone compounds have been shown to possess potent anticancer activities, as well. Small polypeptides that are capable of penetrating the cell membrane and block nuclear translocation of NF-κB is another approach to inhibit its activation. SN50 is a 41 residue synthetic peptide. Its potent effects in blocking the nuclear translocation of NF-κB have shown significant promise as an anticancer agent. However, SN50 also blocks nuclear translocation of other transcription factors (84), thus obscuring the exact contribution of NF-κB blockade to its anticancer effects, and to its potential for manifesting side effects. Several other reports have shown that novel compounds, such as o, o′-bismyristoyl thiamine disulfide, and dehydroxymethylepoxy- quinomicin (85,86) have also been reported to be specific inhibitors of NF-κB nuclear translocation. These are currently being tested as anticancer agents. Although not well investigated, alternate mechanisms are available to suppress NF-κB binding to its cognate DNA element, such as the use of specific methyl transferases, to methylate these sequences and by inhibition of p65 acetylation using specific acetyl transferases.
As illustrated above, NF-κB inhibitors can suppress its activation by different mechanisms. They are being extensively studied in diverse human cancers. However, whether these inhibitors can result in therapeutic benefits in the clinic is still controversial. Long-term suppression of NF-κB activation via these inhibitors will probably induce a homeostatic switch and prevent its efficacy in therapy-resistant tumors. In addition, prophylactic therapy using these inhibitors should also take into account the adverse effects of these agents and how they may result in imbalance of physiological functions in normal cells and tissues. Therefore, it is currently envisioned that the strategies toward safe and efficacious use of these NF-κB pathway inhibitors should be tailored to the patient's genetic background and according to the subtype of tumors that are being targeted. Using a panel of thyroid cancer lines harboring different activating mutations, including the HRAS G13R mutation (C643), BRAFV600E mutations (BCPAR, SW1736), and RET/PTC1 rearrangement (TPC1), Bauerle et al. (43) monitored the antiproliferative effects of a selective genetic inhibitor of NF-κB, a mutant IκBα. Interestingly, the responses to this NF-κB inhibitor were quite different in different cell lines, and on their inhibitory effects on proliferation, apoptosis, and invasion capabilities. These disparate results were believed due to different genetic backgrounds in thyroid cell lines, which represented the potential of combinational strategies to treat thyroid cancer. Most of the activating mutations in thyroid cancer, such as in the RAS, RET, and BRAF genes, are involved in the MAPK signal pathway, which is crucial for tumor initiation (87 –89). Studies have shown that NF-κB activation is involved in the development of resistance to either MAPK or BRAF inhibitors, but the mechanism remains unclear (90 –94). Interestingly, mutant BRAF has been shown to activate NF-κB in melanoma cells by enhancing the ubiquitination and degradation of IκB (95). Liu and Xing (96) had explored the efficacy of dually targeting the NF-κB and MAPK pathways in thyroid cancer. A recent study showed that bortezomib induces apoptosis and growth suppression in human medulloblastoma cells, associated with inhibition of AKT and NF-κB signaling, and synergizes with an ERK inhibitor (97). Studies in thyroid cancer cells suggested that BRAF activates NF-κB and this pathway is MEK-independent (32). Therefore, cotargeting of both MAPK and NF-κB pathways should be effective in suppressing growth of thyroid tumors harboring BRAFV600E mutations, while abrogating resistance due to inhibition of the MEK/ERK pathway. Therefore, it has been postulated that in future studies NF-κB inhibitors should be used in combination with drugs targeting already known abnormalities in parallel signaling pathways, and as an adjunct to cytotoxic chemotherapy or immunotherapy. Multikinase inhibitors, such as Lenvatinib, motesanib, sorafenib, sunitinib, and vandetanib, that can target RET, VEGFR, and other kinases, are studied in clinical trials (3).
Drugs studied as NF-κB inhibitors in thyroid cancer
The underlying mechanisms linked to NF-κB activation and the different inhibitory strategies illustrated above provide the rationale for new therapeutic approaches to treat aggressive thyroid cancers. Some of these inhibitors, such as the proteosomal inhibitor bortezomib, have been clinically approved for multiple myeloma and mantle cell lymphoma. Many other NF-κB inhibitors are also in phase I/II clinical trials for different types of cancers (98,99). However, these NF-κB inhibitors have not been tested for the treatment of thyroid cancers, and no ongoing clinical trials against thyroid cancer are under way even with approved NF-κB inhibitors. Several recent studies using drugs that are able to inhibit the NF-κB pathway in thyroid cancer are mostly in the experimental phase, and data generated have only shown in vitro effects, and few studies showed in vivo effects (43,100 –102). However, it should also be noted that the in vivo targeting of cancer cells by therapeutic drugs is a complex process that involves proper documentation of pharmacokinetic and pharmacodynamic efficacies of these agents. Furthermore, the efficacy of NF-κB inhibitors may only be achieved through simultaneous targeting of multiple pathways. At present, several drugs that have been tested for their antitumor effects in poorly differentiated and anaplastic thyroid cancers include, PS-1145, Bay-11-7082, IKK inhibitor VII, CDDO-Me, and bortezomib, a small-molecule triptolide. Bauerle et al. (43) had used three NF-κB inhibitors, that is, Bay-11-7082, IKK inhibitor VII, and CDDO-Me in a panel of advanced thyroid cancer lines, only one of which showed a significant decrease in cell growth, which was shown to occur via the inhibition of the S-G2/M transition in these cells. Two out of the five thyroid cell lines, SW1736 and TPC1, were sensitive to inhibition of NF-κB by a dominant-negative IκBα (mIκBα), which is resistant to IKK-induced phosphorylation and proteasomal degradation. Studies also showed that triptolide, a small molecule from a Chinese herb, can inhibit the NF-κB activity via blocking the association of p65 with CBP/p300 in the early stage and decreasing the protein level of p65 in the late stage in a human ATC cell line (101,103).
Out of all the NF-κB inhibitors tested so far, only bortezomib was shown to induce significant apoptosis in anaplastic thyroid cancer cell lines. However, this may be due to the effects of bortezomib in targeting numerous other molecules in the cell, such as p21, p27, and BCL-2 family members, as well as several parallel signaling pathways, such as JNK and P53 (100,104). Two recent reports suggested that bortezomib may be a promising agent to treat thyroid cancers (100,102). Therefore, in light of the findings that the NF-κB pathway may play an important role in aggressive thyroid cancers and especially in certain therapy-resistant subtypes which harbor multiple mutations, there needs to be a concerted and systemic effort toward preclinical and ultimately clinical studies to investigate the efficacy of NF-κB inhibitors.
Conclusions
Current investigations clearly suggest that the inhibition of the NF-κB pathway may be a promising strategy for the treatment of advanced thyroid cancers. Despite bortezomib's approval for multiple myeloma and mantle cell lymphoma, there are no inhibitors of NF-κB clinically approved for treatment of thyroid cancer. The NF-κB inhibitors should base their antitumor efficacies by targeting this specific pathway in specific types of thyroid tumors, while avoiding the risk of unexpected side effects. One should also consider that global and prolonged inhibition of NF-κB could manifest detrimental effects to the immune system of the patient. Thus, to avoid long-term immunosuppression and achieve efficacy of NF-κB inhibition, the dosage and schedule for administration of these inhibitors should be carefully considered. Further investigation is needed to develop effective and clinically safe NF-κB inhibitors against aggressive and therapy-resistant thyroid cancers.
Footnotes
Acknowledgments
The authors wish to acknowledge funding from the National Natural Science Foundation of China (no. 30600601), China, and funds from the Tulane Cancer Center (TCC) and Louisiana Cancer Research Consortium (LCRC).
Disclosure Statement
No competing financial interests exist.