
Abstract
Background:
NKX2-1 mutations have been described in several patients with primary congenital hypothyroidism, respiratory distress, and benign hereditary chorea, which are classical manifestations of the brain–thyroid–lung syndrome (BTLS).
Methods:
The NKX2-1 gene was sequenced in the members of a Brazilian family with clinical features of BTLS, and a novel monoallelic mutation was identified in the affected patients. We introduced the mutation in an expression vector for the functional characterization by transfection experiments using both thyroidal and lung-specific promoters.
Results:
The mutation is a deletion of a cytosine at position 834 (ref. sequence NM_003317) (c.493delC) that causes a frameshift with formation of an abnormal protein from amino acid 165 and a premature stop at position 196. The last amino acid of the nuclear localization signal, the whole homeodomain, and the carboxy-terminus of NKX2-1 are all missing in the mutant protein, which has a premature stop codon at position 196 (p.Arg165Glyfs*32). The p.Arg165Glyfs*32 mutant does not bind DNA, and it is unable to transactivate the thyroglobulin (Tg) and the surfactant protein-C (SP-C) promoters. Interestingly, a dose-dependent dominant negative effect of the p.Arg165Glyfs*32 was demonstrated only on the Tg promoter, but not on the SP-C promoter. This effect was also noticed when the mutation was tested in presence of PAX8 or cofactors that synergize with NKX2-1 (P300 and TAZ). The functional effect was also compared with the data present in the literature and demonstrated that, so far, it is very difficult to establish a specific correlation among NKX2-1 mutations, their functional consequence, and the clinical phenotype of affected patients, thus suggesting that the detailed mechanisms of transcriptional regulation still remain unclear.
Conclusions:
We describe a novel NKX2-1 mutation and demonstrate that haploinsufficiency may not be the only explanation for BTLS. Our results indicate that NKX2-1 activity is also finely regulated in a tissue-specific manner, and additional studies are required to better understand the complexities of genotype–phenotype correlations in the NKX2-1 deficiency syndrome.
Introduction
The association of choreoathetosis, hypothyroidism, and pulmonary alterations was linked to mutations in the gene encoding NKX2-1 in the so-called brain–thyroid–lung syndrome (BTLS).
A novel NKX2-1 mutation was identified and functionally characterized in a family with five affected patients in two generations showing hypothyroidism, benign hereditary chorea, and respiratory distress. All the affected members presented a heterozygous deletion of the cytosine at position 834 (ref. sequence NM_003317) of the NKX2-1 gene (c.493delC). The mutation is responsible for a frameshift that produces an abnormal protein from amino acid 165 with a premature stop codon at position 196.
Materials and Methods
Patients
The family included five affected patients in two generations living in Curitiba, Brazil (Fig. 1). All family members gave their informed consent for the study. The study was approved by the ethical committee of each participating institution. The clinical characteristics of the affected members of the family are reported in the Results section.
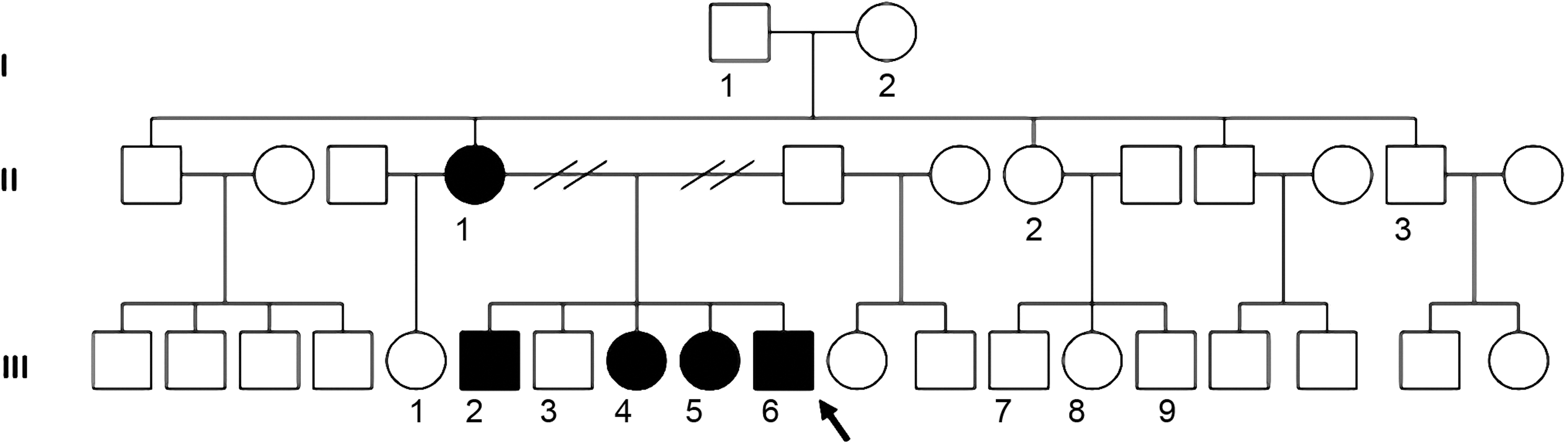
Pedigree of the studied family. The family members carrying the c.493delC mutation (p.Arg165Glyfs*32) and showing the clinical manifestation of brain–thyroid–lung syndrome (BTLS) are in black. The proband (III-6) is indicated by an arrow.
DNA amplification and sequencing
Genomic DNA was extracted from peripheral blood leukocytes of all family members. The three exons of the NKX2-1 gene were amplified by polymerase chain reaction (PCR) as previously described (5). PCR products were purified with Antarctic phosphatase-exonuclease I (New England BioLabs, Ipswich, MA) at 37°C for 15 minutes and 80°C for 15 minutes for the enzyme activation and inactivation respectively and the products were bidirectionally sequenced with a 377 ABI sequencer (Applied Biosystems, Foster City, CA). The genes encoding PAX8 (6), NKX2.5 (7), and TAZ (8) were also amplified and sequenced as previously described. To exclude the presence of polymorphysms, 100 normal subjects were also screened as controls.
Mutagenesis
The mouse Nkx2-1 gene from the p3XFLAG-CMV-10-NKX2-1 vector was used as template in a PCR with the sense primer 5′-CCG CCG GAA GCG CGG GTG CTC TTC T-3′ and the antisense primer 5′-GAG AAG AGC ACC CGC GCT TCC GGC-3′ using the QuikChange Site-Directed Mutagenesis kit (Agilent Technologies, La Jolla, CA) according to the manufacturer's procedure. The presence of the mutation in the obtained vector was confirmed by direct sequencing.
Western blot
HEK293 cells were transiently transfected with 10 μg of either wild-type (WT)-Nkx2-1 or c.493delC-Nkx2-1 plasmids using the FuGene 6 Transfection Reagent (Roche, Indianapolis, IN) according to the manufacturer's instructions. After 48 hours, cells were scraped in phosphate-buffered saline (pH 7.4) and centrifuged at 1792 g for 5 minutes at 4°C. Pellets were frozen and stored at −80°C until used to prepare nuclear or total extracts as previously described (5).
Ten micrograms and 40 μg each of nuclear and total extracts were boiled in Laemmli buffer and resolved on a 12% SDS-PAGE gel. The gel was blotted on Immobilon P (Millipore Corp., Bedford, MA) for 90 minutes at a constant current of 100 mA. Immunodetection of NKX2-1 was performed by using a monoclonal anti-FLAG antibody (M2; Sigma, St. Louis, MO) diluted 1:3000 in Tris-buffered saline containing 5% nonfat milk (Bio-Rad Laboratories, Inc., Richmond, CA), and the filter was treated with a 1:3000 dilution of goat antimouse IgG conjugated to horse radish peroxidase (Bio-Rad).
Electrophoretic mobility shift assay
The binding to DNA of the c.493delC mutant was studied by electrophoretic mobility shift assay (EMSA) using double-stranded end-labeled oligonucleotide C for the Tg promoter as described (9), and double-stranded end-labeled oligonucleotide C2 for the surfactant protein C (SP-C) promoter (10).
Cell culture and in vitro functional assays
HEK293 and HeLa cells were maintained in Dulbecco's modified Eagle's medium supplemented with 2 mM L-glutamine, 4.5 g/L D-glucose, 50 U/mL penicillin, 50 μg/mL streptomycin, and 10% fetal bovine serum under humidified 5% carbon dioxide/95% air at 37°C and grown in 24-well plates to 70%–80% confluence (i.e., ∼5×104 cells per well).
HeLa cells were transfected with the reporter plasmid (0.1 μg of pTg-Luc, 0.3 μg of SP-C-Luc 0.32) plus 3 ng of CMV-Ren as an internal control vector, and different doses of effector plasmids. HEK293 cells were transfected with 0.1 μg of the reporter pTg-Luc plasmid and 1.5 ng of CMV-Ren internal control vector plus different doses of effector plasmids. In each set of transfection experiments, the total amount of transfected DNA was normalized using an empty vector.
Transfections were performed with the TurboFect in vitro Transfection Reagent (Fermentas Life Sciences, Pittsburgh, PA), using a DNA/Turbofect ratio of 1:2 in all experiments. Cells were harvested after 36–48 hours and analyzed sequentially for firefly and Renilla luciferase activities (Dual-Luciferase Reporter Assay System, Promega, Madison, WI). The ratios between the measured firefly and Renilla luciferase activities were expressed relative to the ratios obtained in cells transfected with the reporter and an empty expression vector (CMV-Flag) only. Transfection experiments were performed in duplicate and repeated at least three times. Data are shown as mean±SD. Statistical analysis was performed using Student's t-test.
Results
Patients
The family included five affected patients in three generations (Fig. 1). Collection of clinical information was difficult because the members of the family live in different towns; therefore, some of the clinical characteristics were missing.
The proband (III-6) was the fifth child of nonconsanguineous parents. He was born at term with a birth weight of 2820 g, but a delay in motor development was soon evident: he took his first steps at 24 months of age. During infancy, the proband showed moderate generalized choreiform movements and cerebellar ataxia that remained stable thereafter, accompanied by several episodes of respiratory insufficiency that were diagnosed either as asthma or pneumonia. Routine blood chemistry and hematology tests and electrocardiograms were normal. Brain MRI, electromyography, and muscle biopsy were also normal.
When the proband was 13 years old, his thyroid function tests (TFTs) revealed subclinical hypothyroidism: serum thyrotropin (TSH) was 9.1 mU/L (normal range 0.35–4.94 mU/L), free thyroxine (FT4) was 1.07 ng/dL (normal range 0.7–1.48 ng/dL) and triiodothyronine (T3) was 127.55 ng/dL (normal range 58–159 ng/dL). Serum antithyroid antibodies tests were negative.
The proband's mother (II-1) presented with short stature, mild generalized choreiform movements since infancy, subclinical hypothyroidism (TSH 8.51 mU/L, FT4 1.07 ng/dL, T3 109.46 ng/dL), and episodes of respiratory insufficiency (diagnosed as asthma) in childhood.
Subject III-2, a brother of the proband, presented with cervical dystonia, moderate generalized choreiform movements, and hypothyroidism (TFT not available) at the age of 22 years.
Subject III-4 (the oldest sister of the proband) was diagnosed with a mild choreiform movement disorder at the age of 17 years. At that time, subclinical hypothyroidism (TSH 8.13 mU/L, FT4 1.1 ng/dL, T3 122.08 ng/dL) was also noticed. She was treated with 25 μg/d of L-thyroxine, which normalized serum TSH levels.
The proband's youngest sister (III-5) presented with moderate generalized choreiform movements, mild cognitive deficit, and subclinical hypothyroidism (TSH 5.43 mU/L, FT4 1.19 ng/dL, T3 126.60 ng/dL) at the age of 14 years.
All the other members in the family (I-1, I-2, II-2, II-3, III-1, III-3, III-7, III-8, III-9) had normal TFTs and normal thyroid morphology on ultrasound examination.
Sequencing
Sequencing of the NKX2-1 coding region of the proband's DNA revealed a new monoallelic deletion of the cytosine at position 834 (accession no. NM_003317.3). The mutation is responsible for a frameshift that produces an abnormal protein from amino acid 165. The last amino acid of the nuclear localization signal (NLS), the whole homeodomain, and the carboxy-terminus of NKX2-1 are all missing in the mutant protein, which has a premature stop codon at position 196 (p.Arg165Glyfs*32). The mutant protein has an approximate molecular weight of 20 kDa.
The same mutation (TRANSNP_1331899076), never described before, was present in all the affected members of the family but not in any of the unaffected members of the family or in the 100 normal controls.
No mutations were detected in the coding sequences of PAX8, NKX2-5, and TAZ genes in the family members or in the 100 normal controls.
Nuclear localization
Since the c.493delC mutation occurs in the codon encoding for the last amino acid of the NLS of NKX2-1, we tested the effect on the nuclear localization. To this aim, HeLa cells were transfected with either the WT-Nkx2-1 or the c.493delC-Nkx2-1. After 24 hours, both nuclear and cytoplasmatic proteins were extracted from transfected cells and Western blot results are shown in Figure 2.
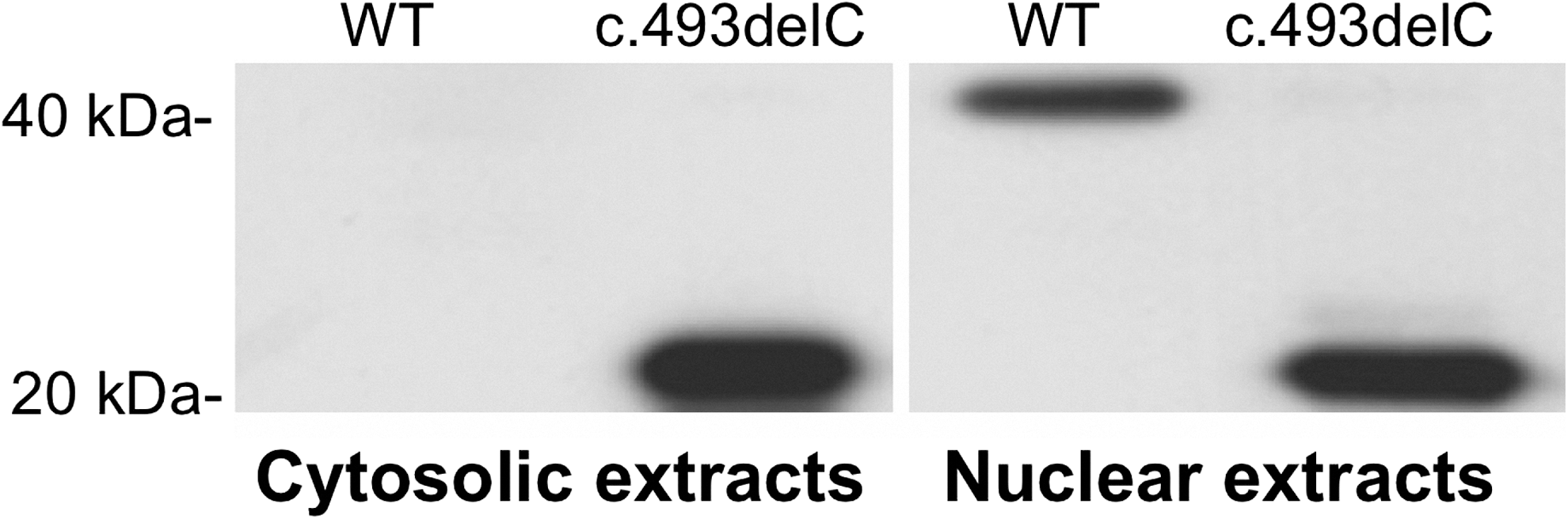
Western blot analysis with the anti-FLAG antibodies of cytosolic and nuclear extract of the cells transfected with both wild-type (WT) and c.493delC (p.Arg165Glyfs*32) cloned into the p3XFLAG-CMV-10 plasmid. The c.493delC is normally synthesized and able to migrate into the nucleus.
As expected, WT-NKX2-1 was synthesized and migrated into the nucleus, disappearing from the cytosolic extracts. Despite the alteration of the nuclear localization signal, the c.493delC was able to migrate into the nucleus.
These results indicate that the p.Arg165Glyfs*32 was synthesized and was capable of migrating into the nucleus.
DNA binding capacity
DNA binding properties of the c.493delC mutant were also analyzed by EMSA using a short DNA stretch corresponding to the NKX2-1 binding site within the Tg promoter (oligo C) (9) and the SP-C promoter (oligo C2) (10). The results are shown in Figure 3. A retarded band could be detected with the WT, while the c.493delC NKX2-1 was, as expected, unable to bind to DNA because of the absence of the homeodomain. Moreover, the addition of the same or double amount of c.493delC extracts did not reduce the binding of WT-NKX2-1. This indicates that c.493delC-NKX2-1 was unable to physically interact and modulate WT-NKX2-1 binding to DNA. The specificity of all the observed complexes was demonstrated in competition experiments adding a 100-fold excess of cold oligos.
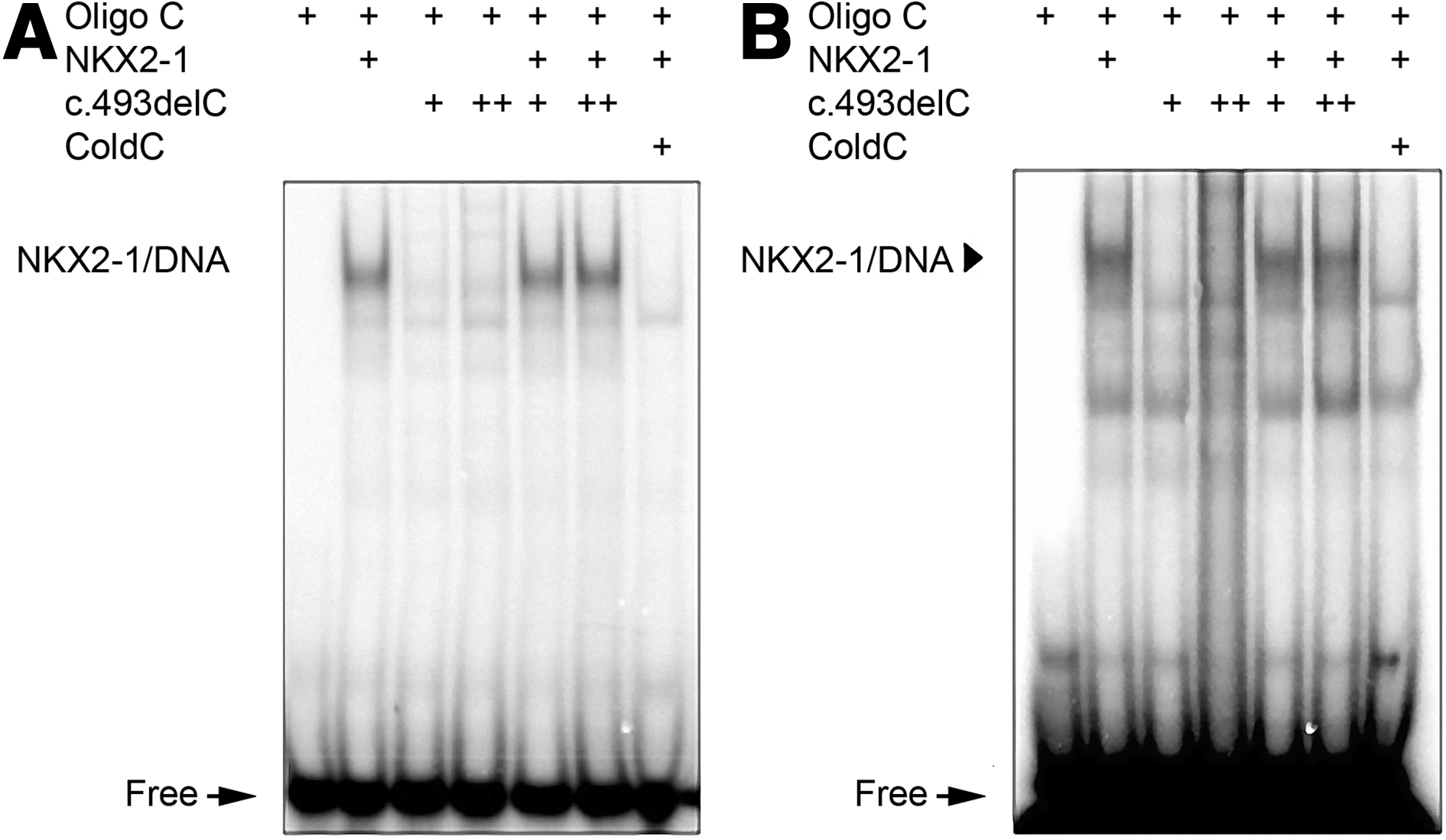
DNA binding capability of the c.493delC mutant (p.Arg165Glyfs*32). Electrophoretic mobility shift assay (EMSA) was performed with nuclear extracts of HEK293 cells transfected with WT and c.493delC constructs on the thyroglobulin (Tg) promoter
Transactivation capacity of mutated and WT NKX2-1 proteins
The transcriptional properties of both WT- and c.493delC-NKX2-1 were investigated by co-transfection assays as previously described. As expected, the c.493delC mutant was unable to activate both the Tg (11) and the SP-C promoters (12).
Thereafter co-transfection experiments were performed using both WT and the c.493delC mutant on the same promoters. Interestingly we were able to demonstrate a dose-dependent dominant negative effect of the c.493delC mutant on the WT-NKX2-1 when used with the Tg promoter (Fig. 4A), but no dominant negative effect could be demonstrated on the SP-C promoter (Fig. 4B), indicating that the c.493delC mutant produced a dominant-negative effect on the wild-type NKX2-1 in a promoter-specific manner.

Transcriptional activation of Tg and the SP-C promoters.
To further understand the dominant-negative action and investigate the c.493delC effects, co-transfection experiments were performed with PAX8, P300, and TAZ on the Tg promoter.
PAX8 (Paired Box Gene 8) is a transcription factor expressed in the thyroid but not in the lung. It synergizes with NKX2-1 to stimulate Tg and thyroperoxidase promoter activities (13).
P300 (E1A-Binding Protein), a general transcriptional coactivator, plays a pivotal role in the activity of many transcription factors, either by bridging sequence-specific DNA-binding factors with elements of the basal transcriptional machinery, such as transcription factor IIB and polymerase 2, or by its intrinsic histone acetyltransferase activity. NKX2-1, PAX8, and P300 physically interact with each other (13 –15), and the functional synergism may involve the direct cooperation of all three factors. The addition of P300 increases the transactivation properties of PAX8, NKX2-1, and both factors together.
TAZ (transcriptional co-activator with a PDZ-binding motif, also known as WWTR1) is also present in thyroid tissue and in differentiated thyroid cell lines (16), thus acting as a potent co-activator for both PAX8 (16) and NKX2-1 (17).
As previously shown (13), we were able to demonstrate that PAX8 synergized with the WT protein and increased Tg promoter activity. The co-transfection with the c.493delC did not modify the PAX8 basal activity, and the dominant negative effect was also present when both WT and c.493delC proteins were transfected with PAX8 (Fig. 5).

Synergism with PAX8. Transcription activation of the Tg promoter by WT-NKX2-1 and c.493delC-NKX2-1 in the interaction with PAX8. HeLa cells have been transfected with 50 ng of each plasmid and 100 ng of the reporter. The addition of WT produced a significant (***p<0.001) increase in the PAX8 activity, while the addition of the c.493delC had no effect. If both WT-Nkx2-1 and c.493delC-Nkx2-1 were transfected, the dominant negative effect of the mutant on the WT protein was still present. The presence of PAX8 did not rescue the dominant negative effect on the Tg promoter.
To further study the interplay of P300, PAX8, and both WT and c.493delC proteins, co-transfection experiments were performed in HEK293 cells, since these cells lack the endogenous P300 (18). Again, if the c.493delC was added to the cells, there was no increase in the activation produced by PAX8 and/or P300, suggesting that this truncated protein did not interfere with the complex. Finally, when all four factors were present (WT, c.493delC, PAX8 and P300), a slight decrease in the activity could be observed compared with the multiprotein complex of NKX2-1/PAX8/P300 (Fig. 6).
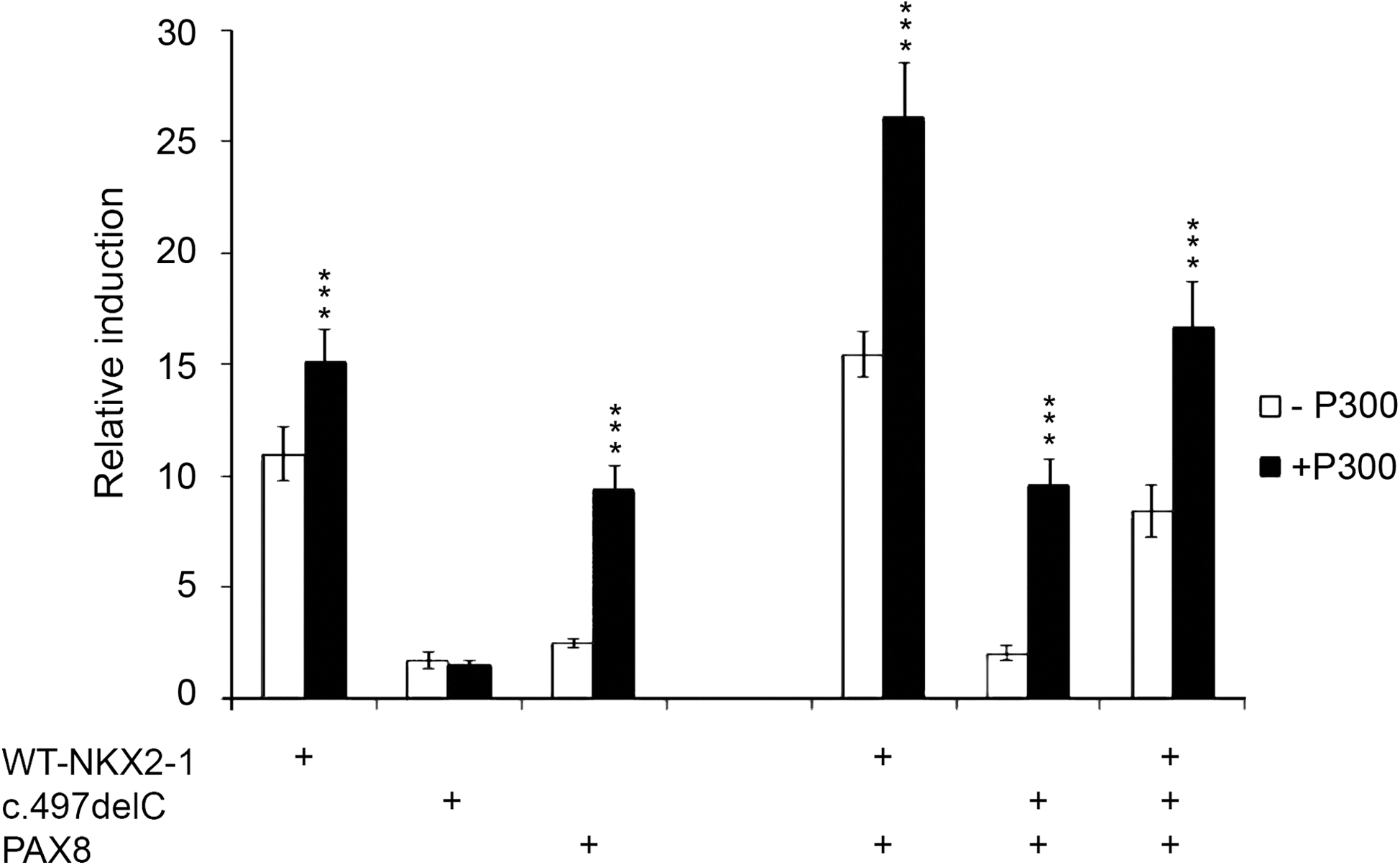
Effect of P300. HEK293 cells were co-transfected with WT-Nkx2-1 (50 ng), c.493delC-Nkx2-1 (50 ng), PAX8 (50 ng), and P300 (300 ng) expression vectors, and the pTg-Luc reporter (100 ng) in the indicated combinations. The addition of P300 significantly increased the activity of the Tg promoter in response to WT-NKX2-1 and PAX8, but had no effects on the activity of c.493delC. The cooperation between WT, PAX8, and P300 produced an additional increase in the activity, while the activation of the complex c.493delC/PAX8 did not differ from the activation observed when PAX8 was co-transfected with P300 alone. Finally, when all the factors were present, there was a reduction of the activation compared with the WT+PAX8 complex, as a consequence of the dominant negative effect of the c.493delC mutant on the WT. Significant differences between the samples with and without P300 are indicated: ***p<0.001.
These results indicate that the dominant negative effect can also be observed when a multifactorial complex is present, and PAX8 or P300 are unable to rescue the effect of the mutation.
Similar results were obtained when the coactivator TAZ was used in HeLa cells (data not shown).
Discussion
Mutations in NKX2-1 have been demonstrated to be responsible for several cases of BTLS, and in this article we describe a novel mutation in the gene encoding for NKX2-1 in a family with the clinical manifestations of benign hereditary chorea, hypothyroidism, and respiratory distress. The mutation c.493delC results in a protein of 196 amino acids. The mutant protein contains a frameshift and differs from the WT beginning at position 165, which corresponds to the last amino acid of the NLS. Transfection experiments with the mutant protein demonstrate that the c.493delC protein is synthesized and translocates into the nucleus, but it is unable to bind DNA since it completely lacks the homeodomain. The c.493delC mutant was responsible for a clear dose-dependent dominant negative effect on the WT NKX2-1 when tested on the Tg promoter, whereas when functional assays were performed on the lung SP-C promoter, the c.493delC mutant showed no dominant negative effect.
Several mutations in NKX2-1 have been reported (5,19 –38), and in the majority of cases haploinsufficiency has been considered to be responsible for the phenotype, as also suggested by the finding of motor abnormalities and mild hyperthyrotropinemia in the Nkx2-1 heterozygous knockout mice (19). Only a few mutations (Table 1) produce a dominant negative effect on the WT NKX2-1 (20,21), and there is only one case among them describing a promoter-specific dominant negative effect (22). In this last case, contrary to what was observed for the c.493delC, the mutant protein contained the homeodomain and could bind to DNA. The authors suggested that the possible interference of the mutant NKX2-1 (p.P275fs*74) with the cooperative interaction between NKX2-1 and PAX8 or NKX2-1 and other cofactors on the Tg promoter is the cause of the promoter-specific dominant negative effect. Since our mutant did not bind to DNA, it is probable that the amino terminus of NKX2-1 binds to other factors that are relevant for its activity on the Tg promoter, thus excluding them from the transcriptional complex and reducing the activity of the WT NKX2-1. In fact, tissue-specific transcriptional regulation is achieved by the combined interaction among transcription factors, co-regulators, and other components regulating basal transcription. In the thyroid, NKX2-1 physically interacts with both PAX8 (13) and TAZ (16). In addition P300, a ubiquitous transcriptional cofactor, has been demonstrated to play an important role for the NKX2-1/PAX8 synergy (14,39). PAX8, P300, and TAZ were tested to see whether they could influence the transactivation of NKX2-1 on its target promoters. The presence of these cofactors increased the activation of the Tg and SP-C promoters, but did not change the thyroid-specific dominant negative effect. In addition, no rescue of the NKX2-1/PAX8 synergism was observed, contrary to what was previously reported in the case of a PAX8 mutation (39).
It has been suggested that NKX2-1 binds to DNA as a dimer (40) and that the interaction with PAX8 occurs via the amino terminus of the NKX2-1 protein (13). Several other factors, most of which not yet defined, may be involved in the regulation of transcription. We believe that the truncated NKX2-1 mutant described in this article interferes with the activity of WT-NKX2-1, decreasing the activity of the multiprotein transcriptional complex. It is likely that PAX8 plays a critical role in modulating this effect in thyroid cells. In lung cells, where PAX8 is absent, the dominant negative effect can not be observed. Even if HeLa cells do not express PAX8, it has been previously suggested that other proteins with a similar activity may replace the function of PAX8 on the Tg promoter (22).
Very recently Silberschmidt and coworkers (41) produced mice lacking one of the two redundant activation domains of Nkx2-1, or having defective phosphorylation of the protein. They demonstrated that each mutant shows a distinct phenotype. They also revealed a level of complexity that could not be predicted by experiments carried out in cultured cells and stressed the discrepancy between the results in the cell models and in those that can be obtained in whole organisms (41). In this context, the major molecular effect of our mutation on the Tg promoter is dominant, while hypothyroidism is only mild (subclinical) in all the affected members of our family. This discrepancy can be the consequence of different mechanisms of action of the mutation in the different tissues, but also of the complex interactions between other genetic and/or environmental factors that may modulate the phenotype. All the reported NKX2-1 mutations (summarized in Table 1) have variable functional effects, even if the mutations occur in similar regions of the protein. Indeed, the clinical features of the affected families and family members described in the literature are very variable, and there is no correlation between the clinical and the molecular phenotype. Mutations that produce major alterations in the protein structure may be associated with only minor neurological signs (19,26), while primary hypothyroidism has been linked to NKX2-1 mutations in 81% of the cases (25/31) (19,20,22,27 –38).
In conclusion, a novel NKX2-1 mutation in a family presenting the clinical findings of the BTLS is described. The c.493delC mutant migrates into the nucleus but is not able to transactivate the Tg or SP-C promoters. These results indicate that NKX2-1 activity is finely regulated by the interaction of several factors that are still largely unknown. The mechanisms regulating gene expression are different in each tissue. Additional studies are required to better understand the complexities of the genotype–phenotype correlation in the NKX2-1 deficiency syndrome.
Footnotes
Acknowledgments
We thank all the members of the affected family for participating in this study. We are grateful to Prof. Jeffrey A. Whitsett (Cincinnati Children's Hospital Medical Center, Cincinnati, OH) for providing us with the human SP-C-Luc plasmid, and to Prof. Mario De Felice (Biogem, Avellino, Italy, and University of Naples “Federico II”) for providing the p3XFLAG-CMV-10-Nkx2-1 vector and for his significant suggestions in the preparation of the manuscript. We also thank Dr. Mariastella Zannini (IEOS-CNR, Naples, Italy) for her helpful discussions and Gloria Tarshes (Naples, Italy) for her contribution in the preparation of this article.
Disclosure Statement
All the authors in the study have no conflict of interest to declare.